Ames Sasha K, Gardner Shea N, Marti Jose Manuel, Slezak Tom R, Gokhale Maya B, Allen Jonathan E
Center for Applied Scientific Computing, Lawrence Livermore National Laboratory, Livermore, California 94550, USA;
Global Security Computer Applications Division, Lawrence Livermore National Laboratory, Livermore, California 94550, USA;
Genome Res. 2015 Jul;25(7):1056-67. doi: 10.1101/gr.184879.114. Epub 2015 Apr 29.
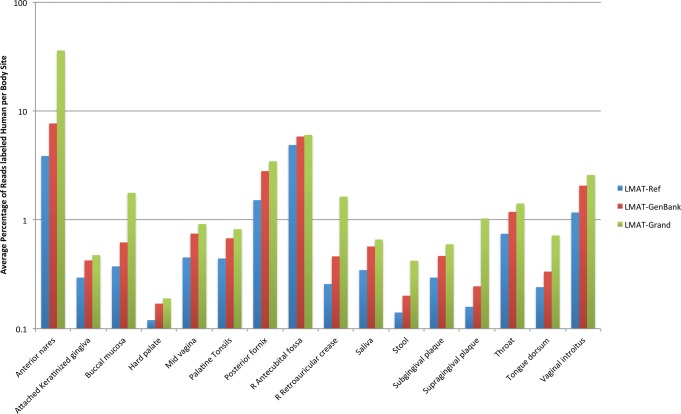
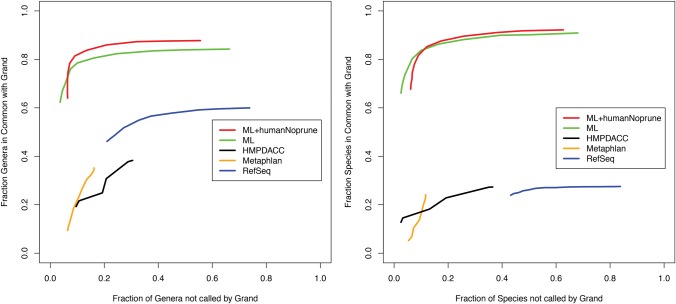
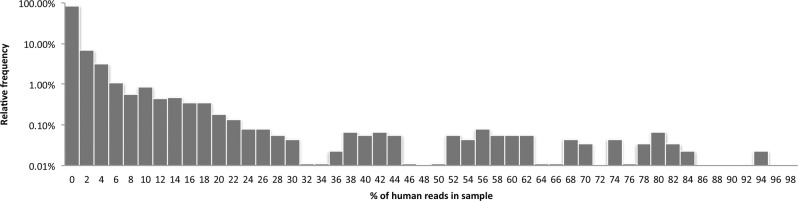
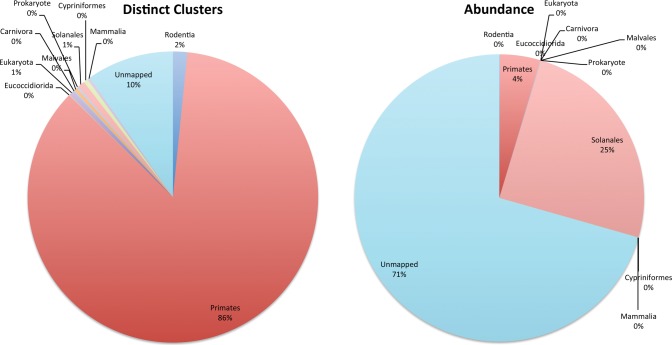
Identifying causative disease agents in human patients from shotgun metagenomic sequencing (SMS) presents a powerful tool to apply when other targeted diagnostics fail. Numerous technical challenges remain, however, before SMS can move beyond the role of research tool. Accurately separating the known and unknown organism content remains difficult, particularly when SMS is applied as a last resort. The true amount of human DNA that remains in a sample after screening against the human reference genome and filtering nonbiological components left from library preparation has previously been underreported. In this study, we create the most comprehensive collection of microbial and reference-free human genetic variation available in a database optimized for efficient metagenomic search by extracting sequences from GenBank and the 1000 Genomes Project. The results reveal new human sequences found in individual Human Microbiome Project (HMP) samples. Individual samples contain up to 95% human sequence, and 4% of the individual HMP samples contain 10% or more human reads. Left unidentified, human reads can complicate and slow down further analysis and lead to inaccurately labeled microbial taxa and ultimately lead to privacy concerns as more human genome data is collected.
当其他靶向诊断方法失效时,通过鸟枪法宏基因组测序(SMS)在人类患者中识别致病病原体是一种强大的工具。然而,在SMS能够超越研究工具的角色之前,仍存在许多技术挑战。准确分离已知和未知的生物体成分仍然很困难,尤其是当SMS作为最后手段应用时。在针对人类参考基因组进行筛选并过滤文库制备过程中留下的非生物成分后,样本中残留的人类DNA的真实数量此前一直未得到充分报道。在本研究中,我们通过从GenBank和千人基因组计划中提取序列,创建了最全面的微生物和无参考人类遗传变异集合,该集合存在于一个为高效宏基因组搜索而优化的数据库中。结果揭示了在人类微生物组计划(HMP)个体样本中发现的新的人类序列。个体样本中人类序列含量高达95%,4%的HMP个体样本中人类读数占比达10%或更多。如果不加以识别,人类读数会使进一步分析变得复杂并减缓分析速度,导致微生物分类群标记不准确,并最终随着更多人类基因组数据的收集引发隐私问题。