Kujur Alice, Upadhyaya Hari D, Shree Tanima, Bajaj Deepak, Das Shouvik, Saxena Maneesha S, Badoni Saurabh, Kumar Vinod, Tripathi Shailesh, Gowda C L L, Sharma Shivali, Singh Sube, Tyagi Akhilesh K, Parida Swarup K
National Institute of Plant Genome Research (NIPGR), Aruna Asaf Ali Marg, New Delhi 110067, India.
International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), Patancheru 502324, Telangana, India.
Sci Rep. 2015 May 5;5:9468. doi: 10.1038/srep09468.
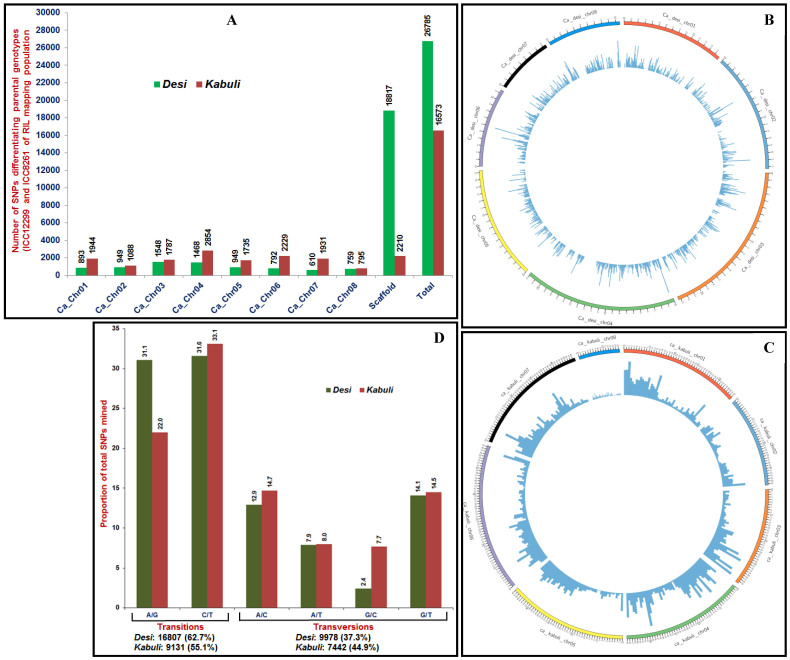
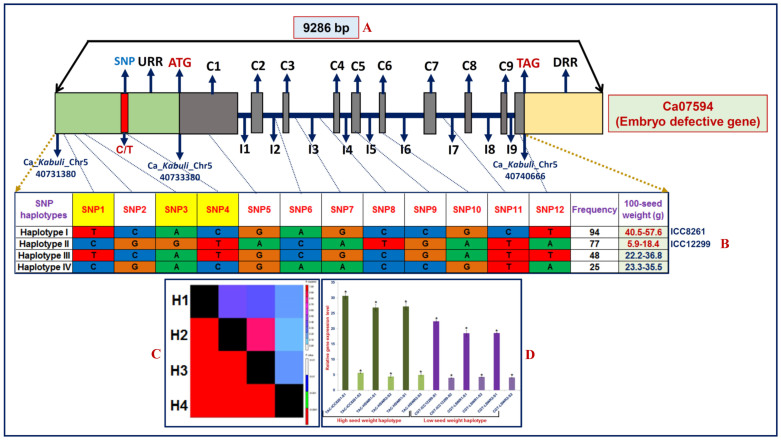
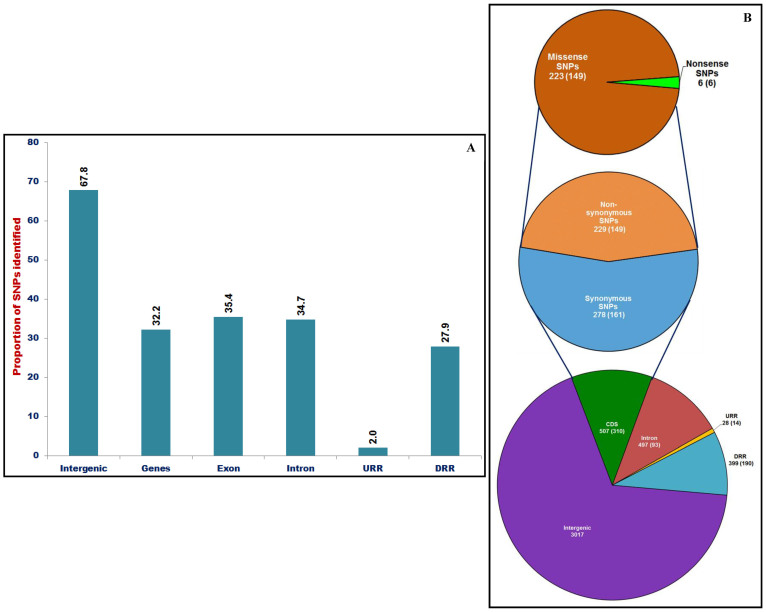
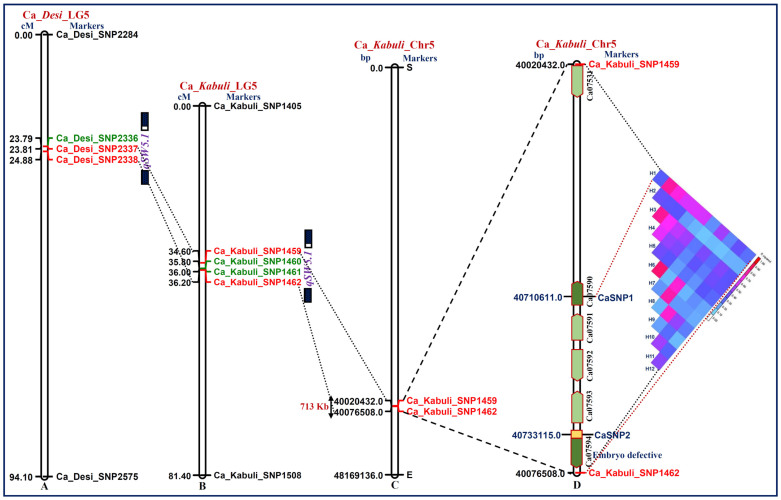
We discovered 26785 and 16573 high-quality SNPs differentiating two parental genotypes of a RIL mapping population using reference desi and kabuli genome-based GBS assay. Of these, 3625 and 2177 SNPs have been integrated into eight desi and kabuli chromosomes, respectively in order to construct ultra-high density (0.20-0.37 cM) intra-specific chickpea genetic linkage maps. One of these constructed high-resolution genetic map has potential to identify 33 major genomic regions harbouring 35 robust QTLs (PVE: 17.9-39.7%) associated with three agronomic traits, which were mapped within <1 cM mean marker intervals on desi chromosomes. The extended LD (linkage disequilibrium) decay (~15 cM) in chromosomes of genetic maps have encouraged us to use a rapid integrated approach (comparative QTL mapping, QTL-region specific haplotype/LD-based trait association analysis, expression profiling and gene haplotype-based association mapping) rather than a traditional QTL map-based cloning method to narrow-down one major seed weight (SW) robust QTL region. It delineated favourable natural allelic variants and superior haplotype-containing one seed-specific candidate embryo defective gene regulating SW in chickpea. The ultra-high-resolution genetic maps, QTLs/genes and alleles/haplotypes-related genomic information generated and integrated strategy for rapid QTL/gene identification developed have potential to expedite genomics-assisted breeding applications in crop plants, including chickpea for their genetic enhancement.
我们利用基于参考迪西(desi)和卡布利(kabuli)基因组的简化基因组测序(GBS)分析,在一个重组自交系(RIL)作图群体的两个亲本基因型中发现了26785个和16573个高质量单核苷酸多态性(SNP)。其中,3625个和2177个SNP分别整合到了8条迪西和卡布利染色体上,以构建超高密度(0.20 - 0.37厘摩)的种内鹰嘴豆遗传连锁图谱。构建的这些高分辨率遗传图谱之一有潜力识别出33个主要基因组区域,这些区域含有与三个农艺性状相关的35个稳定的数量性状基因座(QTL,表型变异解释率:17.9 - 39.7%),这些QTL被定位在迪西染色体上平均标记间隔小于1厘摩的范围内。遗传图谱染色体中较长的连锁不平衡(LD)衰退(约15厘摩)促使我们采用一种快速综合方法(比较QTL定位、基于QTL区域特定单倍型/连锁不平衡的性状关联分析、表达谱分析和基于基因单倍型的关联作图),而不是传统的基于QTL图谱的克隆方法,来缩小一个主要种子重量(SW)稳定QTL区域。它描绘了有利的自然等位基因变异和包含一个种子特异性候选胚胎缺陷基因的优良单倍型,该基因调控鹰嘴豆的种子重量。所生成的超高分辨率遗传图谱、与QTL/基因以及等位基因/单倍型相关的基因组信息,以及所开发的用于快速QTL/基因鉴定的综合策略,有潜力加速包括鹰嘴豆在内的作物植物的基因组辅助育种应用,以实现其遗传改良。