Saxena Maneesha S, Bajaj Deepak, Das Shouvik, Kujur Alice, Kumar Vinod, Singh Mohar, Bansal Kailash C, Tyagi Akhilesh K, Parida Swarup K
National Institute of Plant Genome Research (NIPGR), Aruna Asaf Ali Marg, New Delhi 110067, India.
National Research Centre on Plant Biotechnology (NRCPB), New Delhi 110012, India.
DNA Res. 2014 Dec;21(6):695-710. doi: 10.1093/dnares/dsu031. Epub 2014 Oct 21.
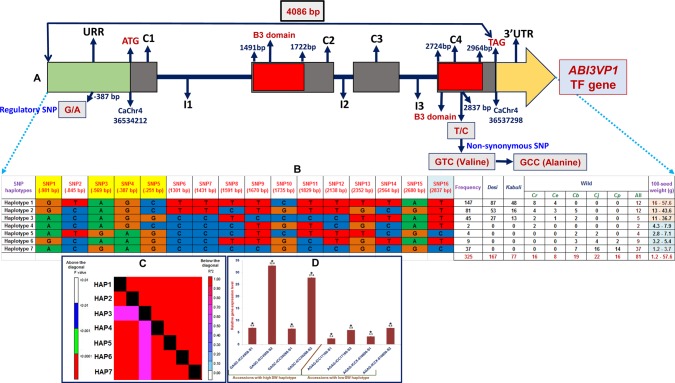
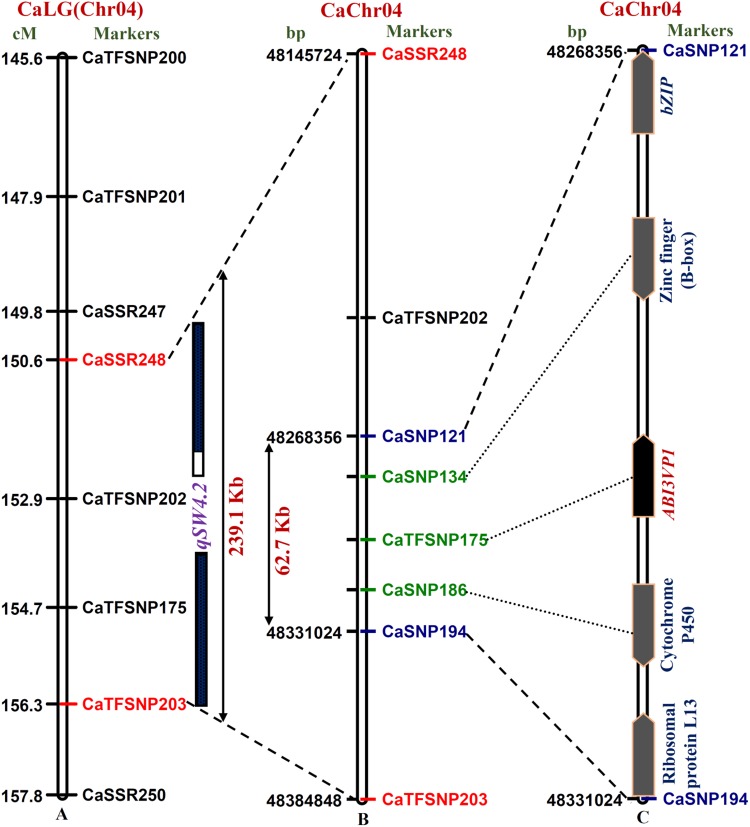
The identification and fine mapping of robust quantitative trait loci (QTLs)/genes governing important agro-morphological traits in chickpea still lacks systematic efforts at a genome-wide scale involving wild Cicer accessions. In this context, an 834 simple sequence repeat and single-nucleotide polymorphism marker-based high-density genetic linkage map between cultivated and wild parental accessions (Cicer arietinum desi cv. ICC 4958 and Cicer reticulatum wild cv. ICC 17160) was constructed. This inter-specific genetic map comprising eight linkage groups spanned a map length of 949.4 cM with an average inter-marker distance of 1.14 cM. Eleven novel major genomic regions harbouring 15 robust QTLs (15.6-39.8% R(2) at 4.2-15.7 logarithm of odds) associated with four agro-morphological traits (100-seed weight, pod and branch number/plant and plant hairiness) were identified and mapped on chickpea chromosomes. Most of these QTLs showed positive additive gene effects with effective allelic contribution from ICC 4958, particularly for increasing seed weight (SW) and pod and branch number. One robust SW-influencing major QTL region (qSW4.2) has been narrowed down by combining QTL mapping with high-resolution QTL region-specific association analysis, differential expression profiling and gene haplotype-based association/LD mapping. This enabled to delineate a strong SW-regulating ABI3VP1 transcription factor (TF) gene at trait-specific QTL interval and consequently identified favourable natural allelic variants and superior high seed weight-specific haplotypes in the upstream regulatory region of this gene showing increased transcript expression during seed development. The genes (TFs) harbouring diverse trait-regulating QTLs, once validated and fine-mapped by our developed rapid integrated genomic approach and through gene/QTL map-based cloning, can be utilized as potential candidates for marker-assisted genetic enhancement of chickpea.
在鹰嘴豆中,对控制重要农艺形态性状的稳健数量性状基因座(QTL)/基因进行鉴定和精细定位,在涉及野生鹰嘴豆种质的全基因组范围内仍缺乏系统的研究。在此背景下,构建了一个基于834个简单序列重复和单核苷酸多态性标记的栽培亲本与野生亲本(栽培鹰嘴豆品种ICC 4958和野生鹰嘴豆品种ICC 17160)之间的高密度遗传连锁图谱。这个包含8个连锁群的种间遗传图谱全长949.4厘摩,标记间平均距离为1.14厘摩。鉴定出11个包含15个稳健QTL的新主要基因组区域(在4.2至15.7的对数优势比下,贡献率为15.6 - 39.8%),这些QTL与四个农艺形态性状(百粒重、每株荚数和分枝数以及植株茸毛)相关,并定位到鹰嘴豆染色体上。这些QTL大多表现出正向加性基因效应,ICC 4958有有效的等位基因贡献,特别是在增加种子重量(SW)以及荚数和分枝数方面。通过将QTL定位与高分辨率QTL区域特异性关联分析、差异表达谱分析以及基于基因单倍型的关联/连锁不平衡定位相结合,一个影响种子重量的稳健主要QTL区域(qSW4.2)已被缩小范围。这使得能够在性状特异性QTL区间确定一个强烈调控种子重量的ABI3VP1转录因子(TF)基因,并因此在该基因的上游调控区域鉴定出有利的自然等位变异和优良的高种子重量特异性单倍型,这些单倍型在种子发育过程中表现出转录表达增加。一旦通过我们开发的快速综合基因组方法以及基于基因/QTL图谱的克隆对包含不同性状调控QTL的基因(TF)进行验证和精细定位,它们就可以用作鹰嘴豆标记辅助遗传改良的潜在候选基因。