Park Hahnbeom, DiMaio Frank, Baker David
Department of Biochemistry, University of Washington, Seattle, WA 98195, USA; Institute for Protein Design, University of Washington, Seattle, WA 98195, USA.
Department of Biochemistry, University of Washington, Seattle, WA 98195, USA; Institute for Protein Design, University of Washington, Seattle, WA 98195, USA; Howard Hughes Medical Institute, University of Washington, Box 357370, Seattle, WA 98195, USA.
Structure. 2015 Jun 2;23(6):1123-8. doi: 10.1016/j.str.2015.03.022. Epub 2015 May 7.
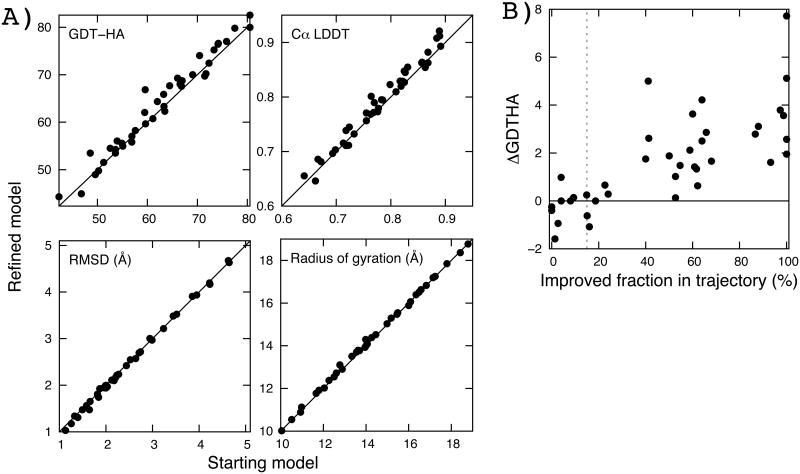
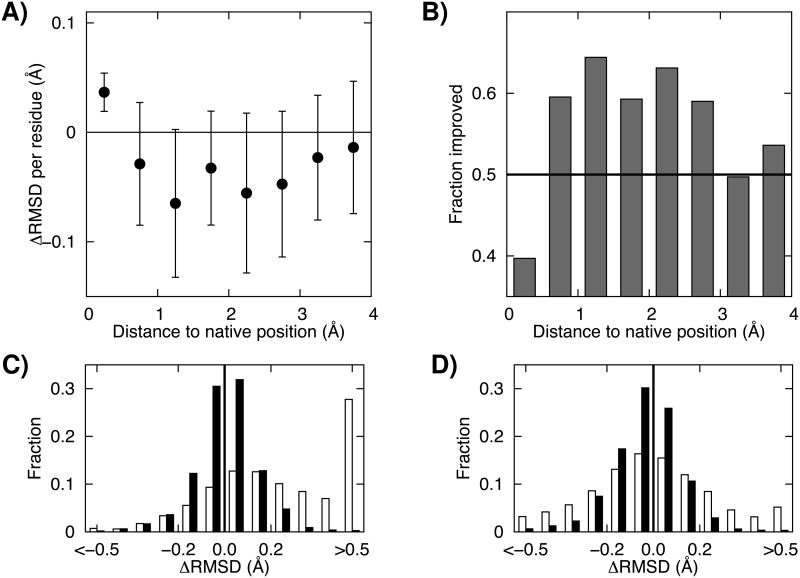
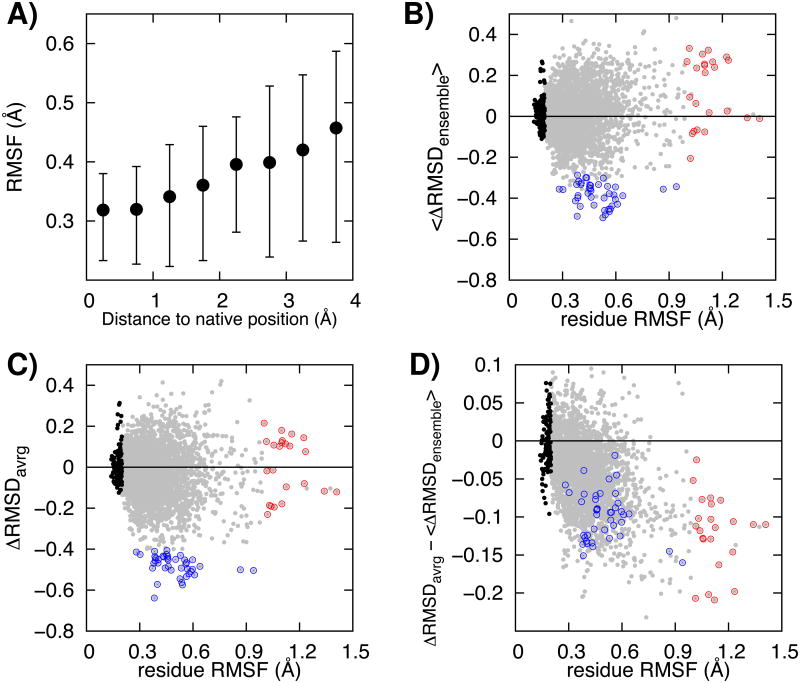
Recent studies have shown that explicit solvent molecular dynamics (MD) simulation followed by structural averaging can consistently improve protein structure models. We find that improvement upon averaging is not limited to explicit water MD simulation, as consistent improvements are also observed for more efficient implicit solvent MD or Monte Carlo minimization simulations. To determine the origin of these improvements, we examine the changes in model accuracy brought about by averaging at the individual residue level. We find that the improvement in model quality from averaging results from the superposition of two effects: a dampening of deviations from the correct structure in the least well modeled regions, and a reinforcement of consistent movements towards the correct structure in better modeled regions. These observations are consistent with an energy landscape model in which the magnitude of the energy gradient toward the native structure decreases with increasing distance from the native state.
最近的研究表明,通过显式溶剂分子动力学(MD)模拟并随后进行结构平均,可以持续改进蛋白质结构模型。我们发现,平均后的改进不仅限于显式水MD模拟,因为在更高效的隐式溶剂MD或蒙特卡罗最小化模拟中也观察到了一致的改进。为了确定这些改进的来源,我们在单个残基水平上研究了平均所带来的模型准确性变化。我们发现,平均后模型质量的提高源于两种效应的叠加:在建模效果最差的区域中,偏差相对于正确结构的减小;在建模效果较好的区域中,向正确结构的一致运动的增强。这些观察结果与能量景观模型一致,在该模型中,朝着天然结构的能量梯度大小随着与天然状态距离的增加而减小。