Illingworth Christopher J R
Department of Genetics, University of Cambridge, Cambridge, United Kingdom
Mol Biol Evol. 2015 Nov;32(11):3012-26. doi: 10.1093/molbev/msv171. Epub 2015 Aug 4.
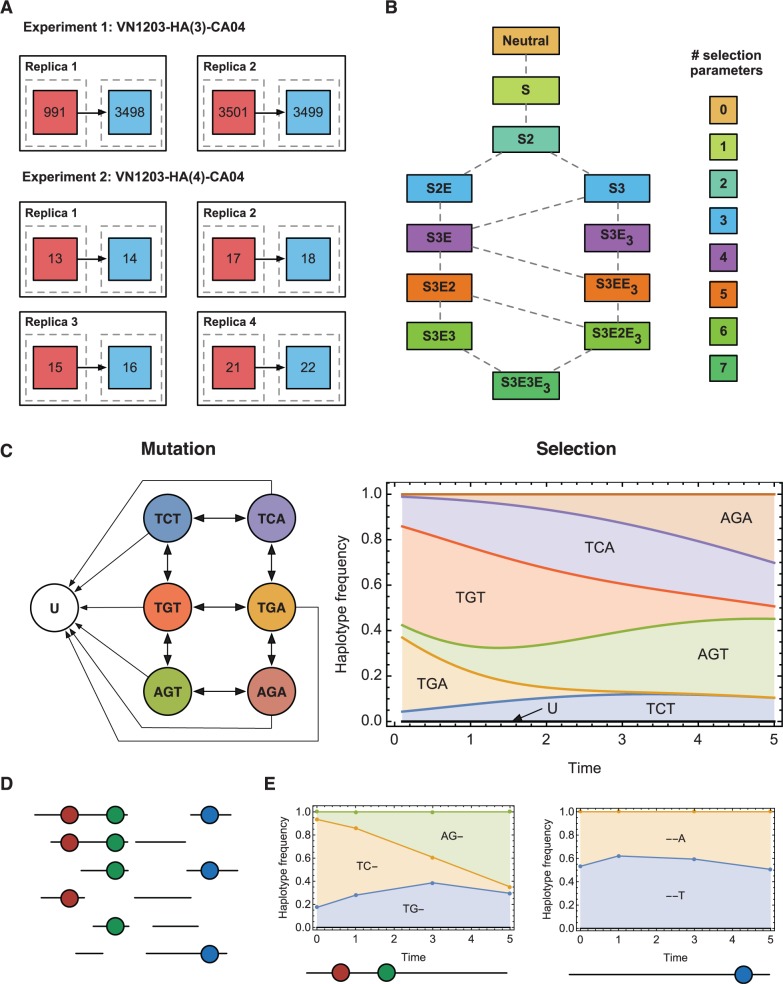
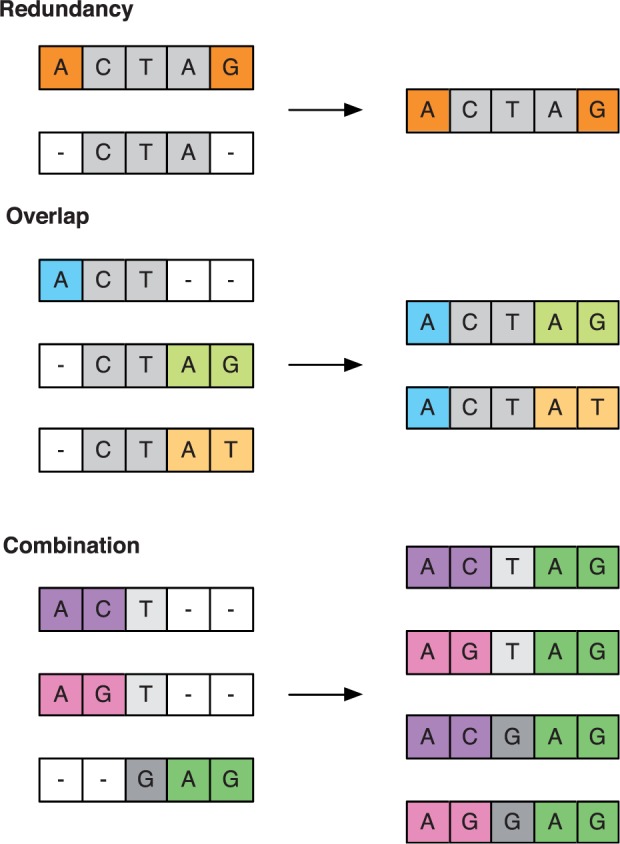
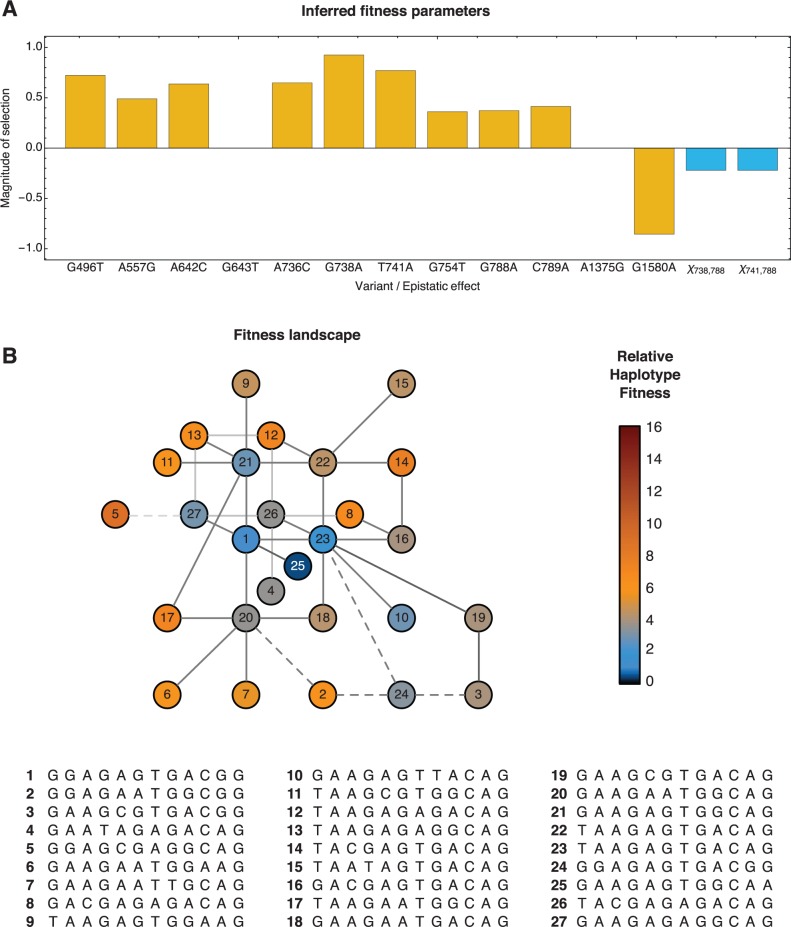
We present a method to infer the role of selection acting during the within-host evolution of the influenza virus from short-read genome sequence data. Linkage disequilibrium between loci is accounted for by treating short-read sequences as noisy multilocus emissions from an underlying model of haplotype evolution. A hierarchical model-selection procedure is used to infer the underlying fitness landscape of the virus insofar as that landscape is explored by the viral population. In a first application of our method, we analyze data from an evolutionary experiment describing the growth of a reassortant H5N1 virus in ferrets. Across two sets of replica experiments we infer multiple alleles to be under selection, including variants associated with receptor binding specificity, glycosylation, and with the increased transmissibility of the virus. We identify epistasis as an important component of the within-host fitness landscape, and show that adaptation can proceed through multiple genetic pathways.
我们提出了一种方法,可从短读长基因组序列数据推断在流感病毒宿主内进化过程中起作用的选择的作用。通过将短读长序列视为单倍型进化潜在模型产生的有噪声的多位点排放,来考虑位点间的连锁不平衡。使用分层模型选择程序来推断病毒的潜在适应度景观,只要该景观是由病毒群体探索的。在我们方法的首次应用中,我们分析了来自一项进化实验的数据,该实验描述了重配H5N1病毒在雪貂体内的生长情况。在两组重复实验中,我们推断多个等位基因处于选择之下,包括与受体结合特异性、糖基化以及病毒传播性增加相关的变体。我们确定上位性是宿主内适应度景观的一个重要组成部分,并表明适应可以通过多种遗传途径进行。