Grassi Elena, Zapparoli Ettore, Molineris Ivan, Provero Paolo
Dept. of Molecular Biotechnology and Health Sciences, University of Turin, Turin, Italy.
Center for Translational Genomics and Bioinformatics, San Raffaele Scientific Institute, Milan, Italy.
PLoS One. 2015 Nov 24;10(11):e0143627. doi: 10.1371/journal.pone.0143627. eCollection 2015.
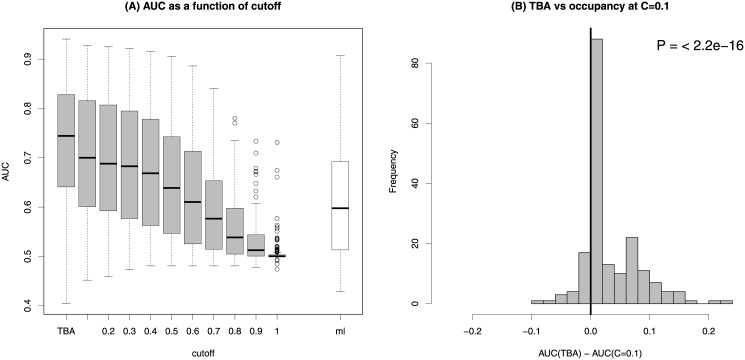
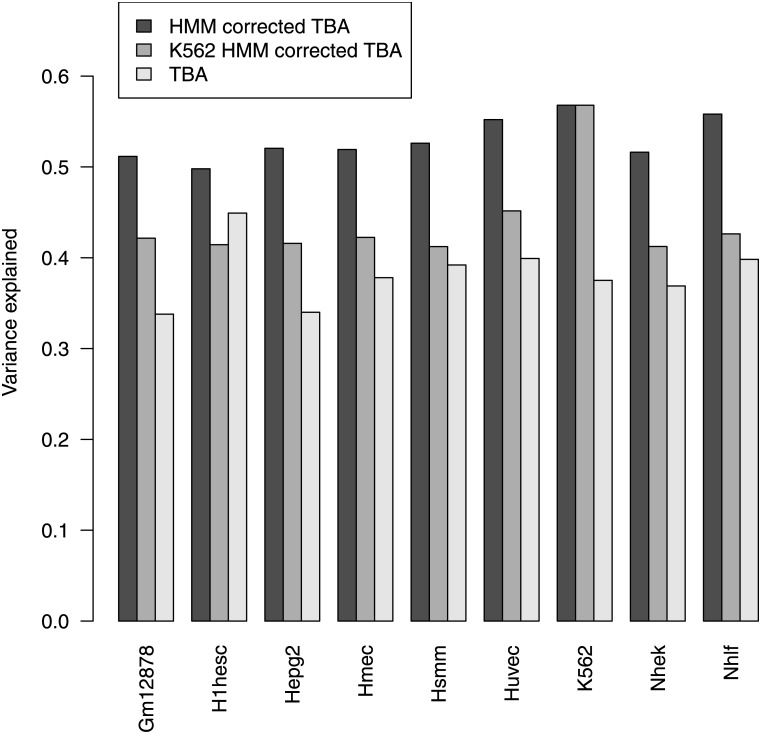
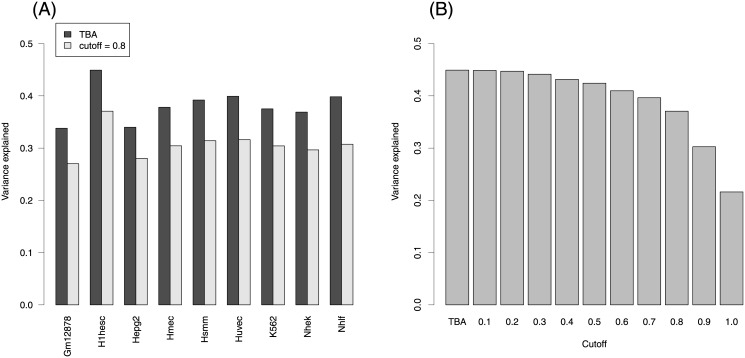
Transcription factors regulate gene expression by binding regulatory DNA. Understanding the rules governing such binding is an essential step in describing the network of regulatory interactions, and its pathological alterations. We show that describing regulatory regions in terms of their profile of total binding affinities for transcription factors leads to increased predictive power compared to methods based on the identification of discrete binding sites. This applies both to the prediction of transcription factor binding as revealed by ChIP-seq experiments and to the prediction of gene expression through RNA-seq. Further significant improvements in predictive power are obtained when regulatory regions are defined based on chromatin states inferred from histone modification data.
转录因子通过结合调控性DNA来调节基因表达。理解此类结合的规则是描述调控相互作用网络及其病理改变的关键一步。我们发现,与基于离散结合位点识别的方法相比,根据转录因子的总结合亲和力谱来描述调控区域可提高预测能力。这既适用于ChIP-seq实验所揭示的转录因子结合预测,也适用于通过RNA-seq对基因表达的预测。当基于从组蛋白修饰数据推断出的染色质状态来定义调控区域时,预测能力会进一步显著提高。