Shan Hong, Wang Zihao, Zhang Fa, Xiong Yong, Yin Chang-Cheng, Sun Fei
Department of Biophysics, College of Basic Medical Sciences, Peking University Health Science Center, Beijing, 100191, China.
Key Lab of Intelligent Information Processing and Advanced Computing Research Lab, Institute of Computing Technology, Chinese Academy of Sciences, Beijing, 100190, China.
Protein Cell. 2016 Jan;7(1):46-62. doi: 10.1007/s13238-015-0229-2. Epub 2015 Dec 17.
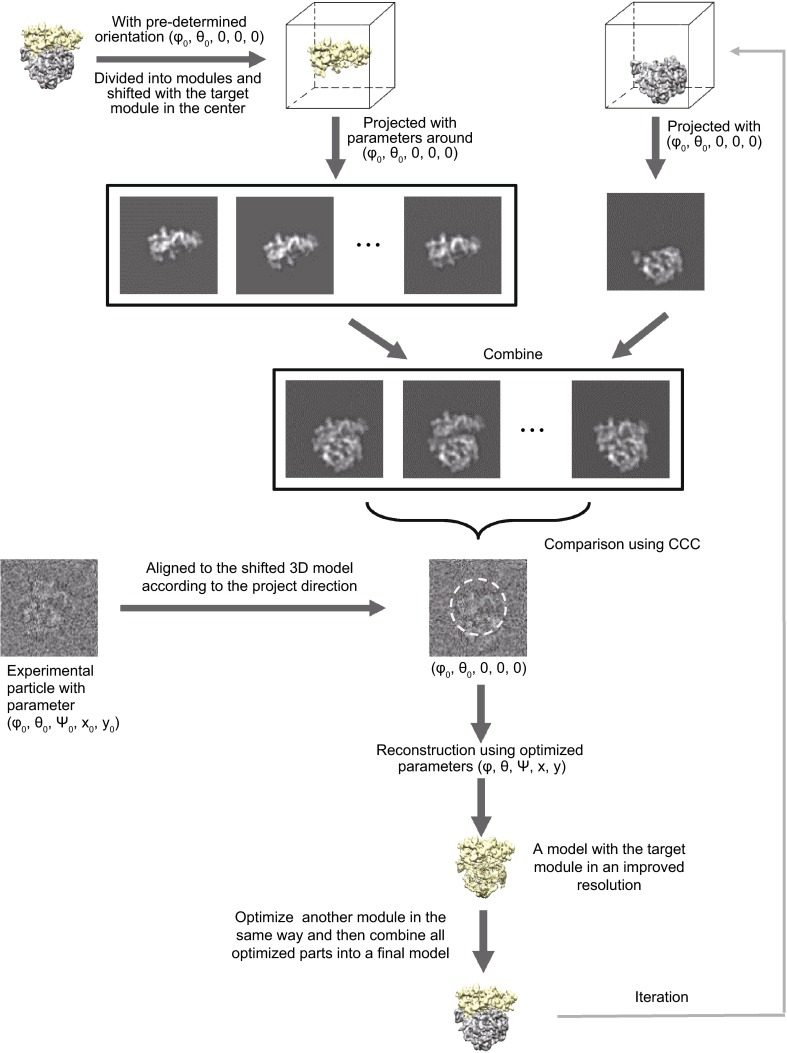
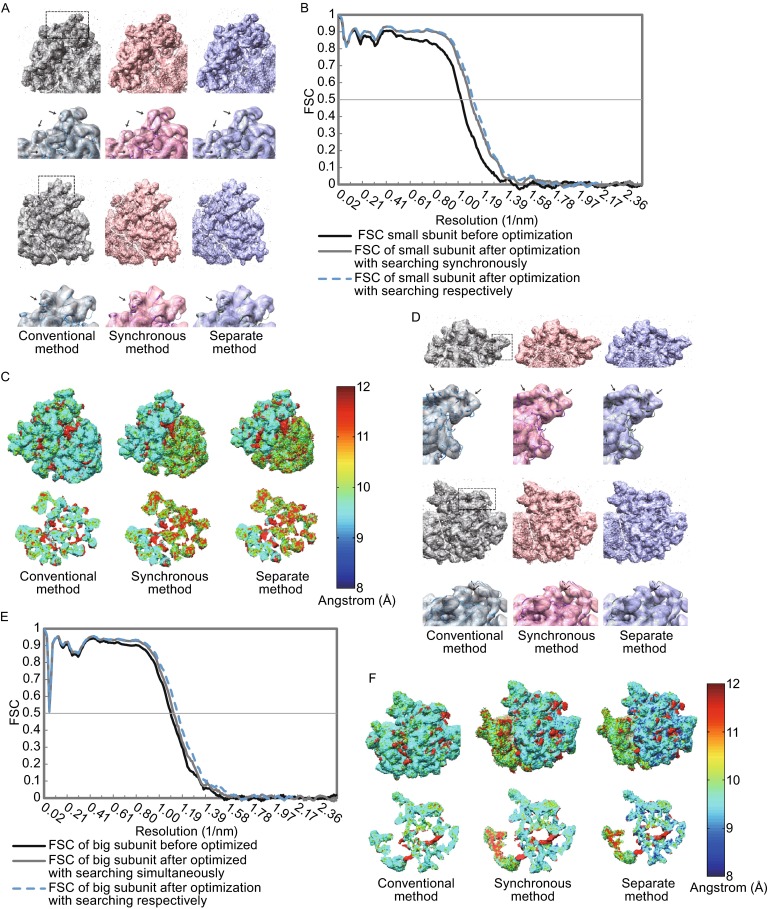
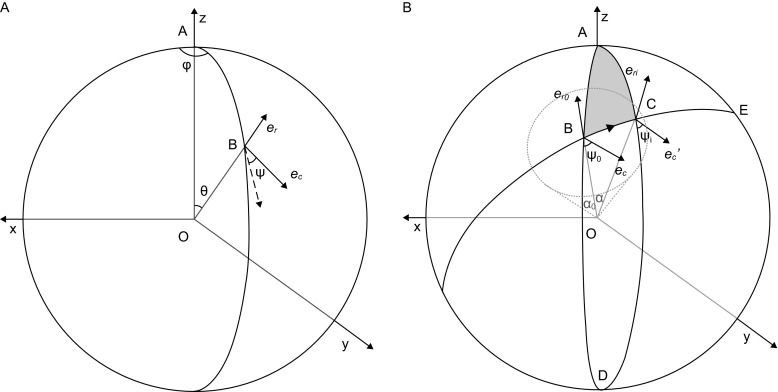
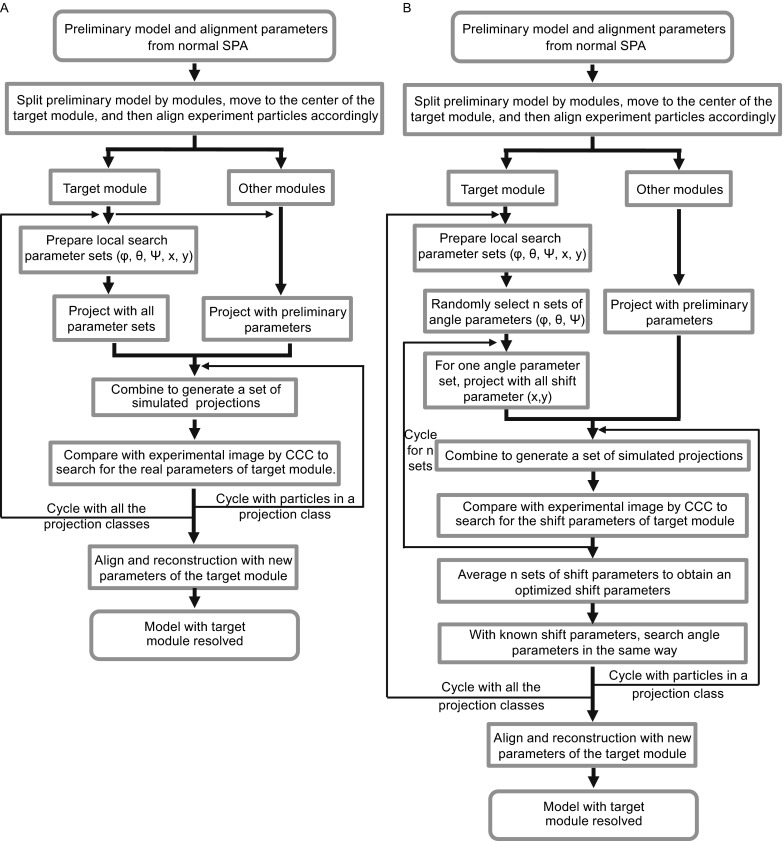
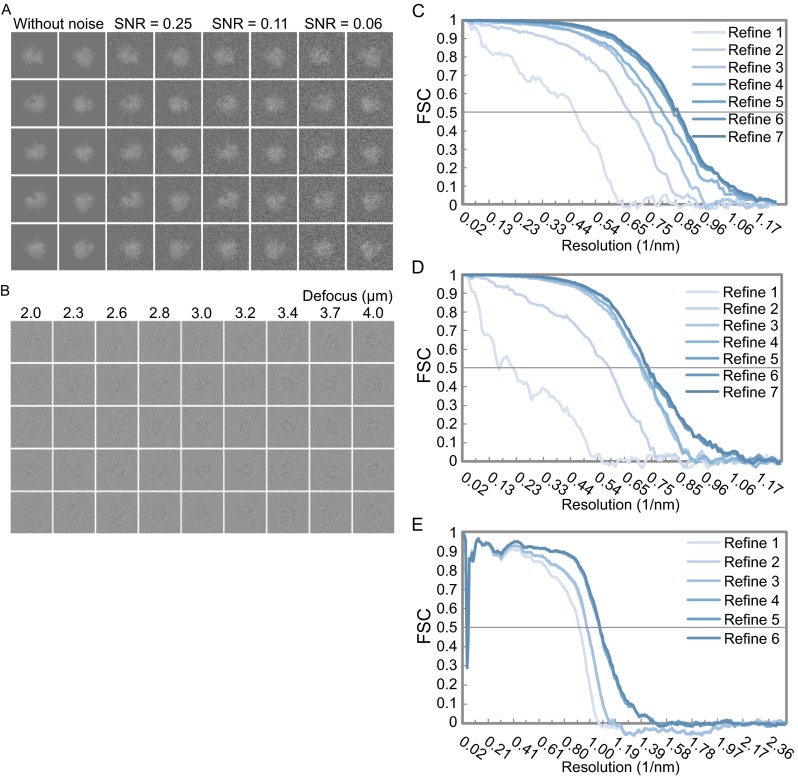
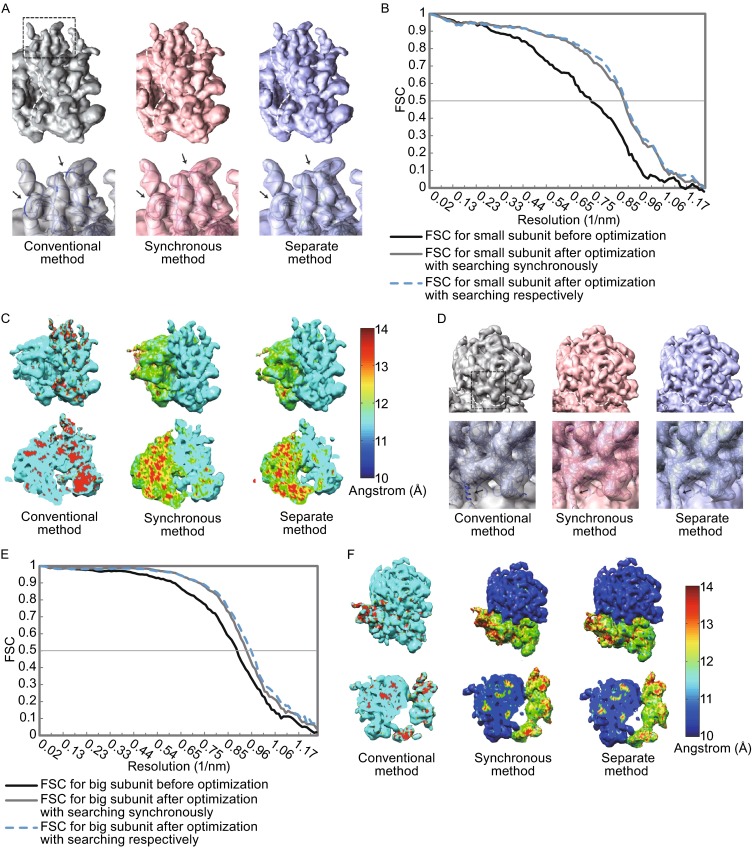
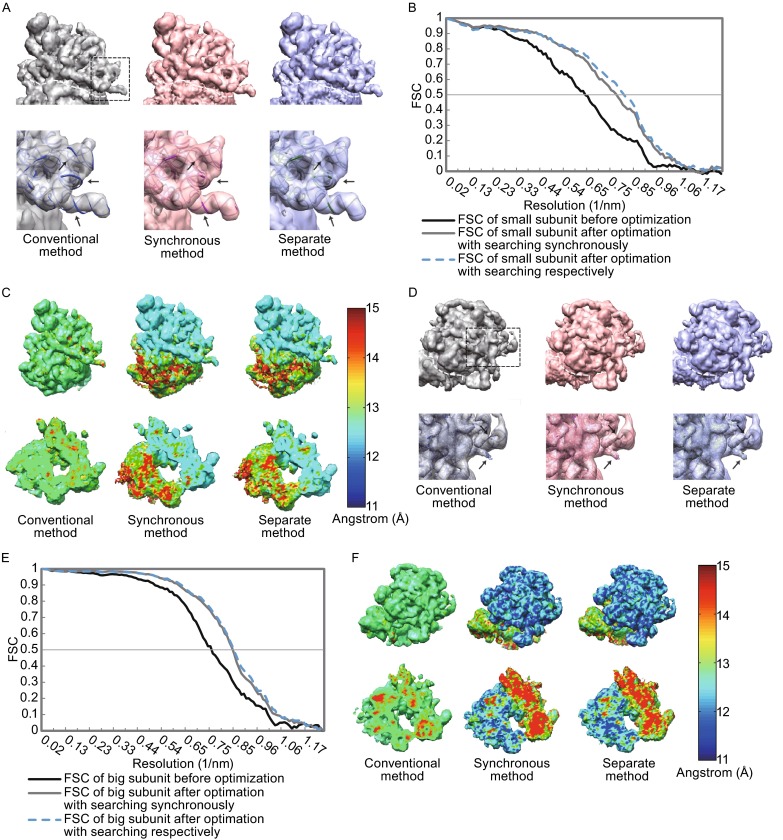
Single particle analysis, which can be regarded as an average of signals from thousands or even millions of particle projections, is an efficient method to study the three-dimensional structures of biological macromolecules. An intrinsic assumption in single particle analysis is that all the analyzed particles must have identical composition and conformation. Thus specimen heterogeneity in either composition or conformation has raised great challenges for high-resolution analysis. For particles with multiple conformations, inaccurate alignments and orientation parameters will yield an averaged map with diminished resolution and smeared density. Besides extensive classification approaches, here based on the assumption that the macromolecular complex is made up of multiple rigid modules whose relative orientations and positions are in slight fluctuation around equilibriums, we propose a new method called as local optimization refinement to address this conformational heterogeneity for an improved resolution. The key idea is to optimize the orientation and shift parameters of each rigid module and then reconstruct their three-dimensional structures individually. Using simulated data of 80S/70S ribosomes with relative fluctuations between the large (60S/50S) and the small (40S/30S) subunits, we tested this algorithm and found that the resolutions of both subunits are significantly improved. Our method provides a proof-of-principle solution for high-resolution single particle analysis of macromolecular complexes with dynamic conformations.
单颗粒分析可被视为来自数千甚至数百万个颗粒投影信号的平均值,是研究生物大分子三维结构的一种有效方法。单颗粒分析中的一个内在假设是,所有被分析的颗粒必须具有相同的组成和构象。因此,组成或构象方面的样本异质性给高分辨率分析带来了巨大挑战。对于具有多种构象的颗粒,不准确的对齐和取向参数将产生分辨率降低且密度模糊的平均图谱。除了广泛的分类方法外,基于大分子复合物由多个刚性模块组成,其相对取向和位置在平衡附近有轻微波动的假设,我们提出了一种称为局部优化细化的新方法来解决这种构象异质性以提高分辨率。关键思想是优化每个刚性模块的取向和位移参数,然后分别重建它们的三维结构。使用大亚基(60S/50S)和小亚基(40S/30S)之间具有相对波动的80S/70S核糖体的模拟数据,我们测试了该算法,发现两个亚基的分辨率都有显著提高。我们的方法为具有动态构象的大分子复合物的高分辨率单颗粒分析提供了一个原理验证解决方案。