Li Dingchen, Li Zongwei, Zhou Zhe, Li Zhen, Qu Xinyan, Xu Peisong, Zhou Pingkun, Bo Xiaochen, Ni Ming
Department of Biotechnology, Beijing Institute of Radiation Medicine, 27 Taiping Road, Beijing, 100850, People's Republic of China.
Genomics Center of Academy of Military Medical Sciences, 27 Taiping Road, Beijing, 100850, People's Republic of China.
Biol Direct. 2016 Jan 12;11(1):3. doi: 10.1186/s13062-016-0105-x.
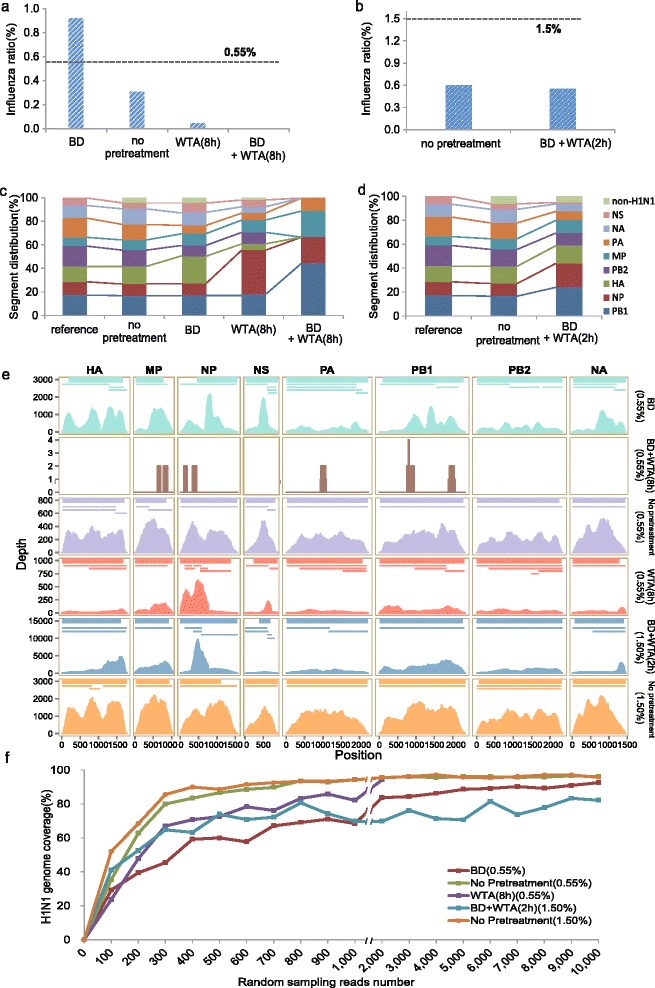
Next-generation sequencing (NGS) enables the recovery of pathogen genomes from clinical samples without the need for culturing. Depletion of host/microbiota components (e.g., ribosomal RNA and poly-A RNA) and whole DNA/cDNA amplification are routine methods to improve recovery results. Using mixtures of human and influenza A virus (H1N1) RNA as a model, we found that background depletion and whole transcriptome amplification introduced biased distributions of read coverage over the H1N1 genome, thereby hampering genome assembly. Influenza serotyping was also affected by pretreatments. We propose that direct sequencing of noncultured samples without pretreatment is a favorable option for pathogen genome recovery applications.
下一代测序(NGS)无需培养即可从临床样本中获取病原体基因组。去除宿主/微生物群成分(如核糖体RNA和多聚腺苷酸RNA)以及全DNA/cDNA扩增是提高获取结果的常规方法。以人类和甲型流感病毒(H1N1)RNA混合物为模型,我们发现背景去除和全转录组扩增会导致H1N1基因组上的读取覆盖分布出现偏差,从而妨碍基因组组装。流感血清分型也受到预处理的影响。我们建议,对于病原体基因组获取应用而言,未经预处理的非培养样本直接测序是一个不错的选择。