Yoshida Kohta, Miyagi Ryutaro, Mori Seiichi, Takahashi Aya, Makino Takashi, Toyoda Atsushi, Fujiyama Asao, Kitano Jun
Division of Ecological Genetics Department of Population Genetics National Institute of Genetics Mishima Shizuoka Japan.
Evolutionary Genetics Laboratory Department of Biological Sciences Tokyo Metropolitan University Hachioji Tokyo Japan.
Ecol Evol. 2016 Mar 2;6(7):2190-204. doi: 10.1002/ece3.2047. eCollection 2016 Apr.
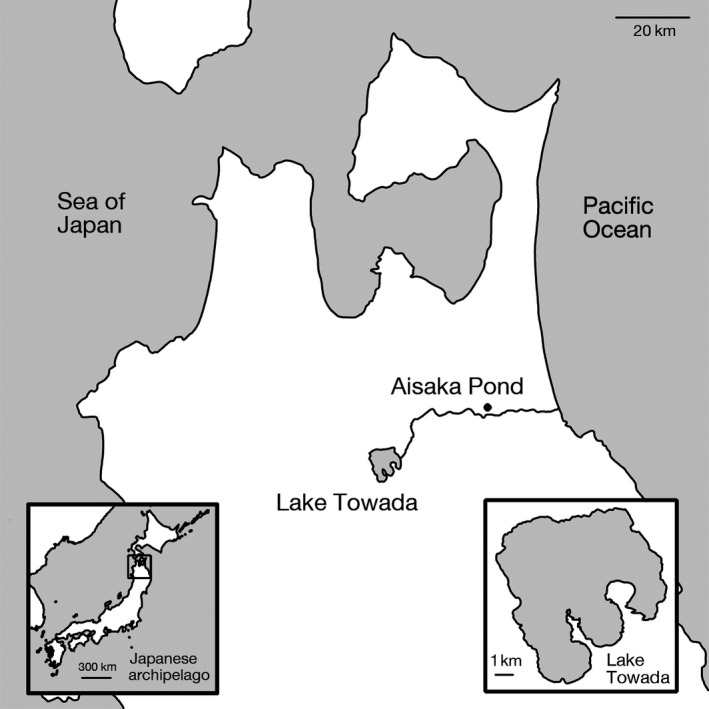
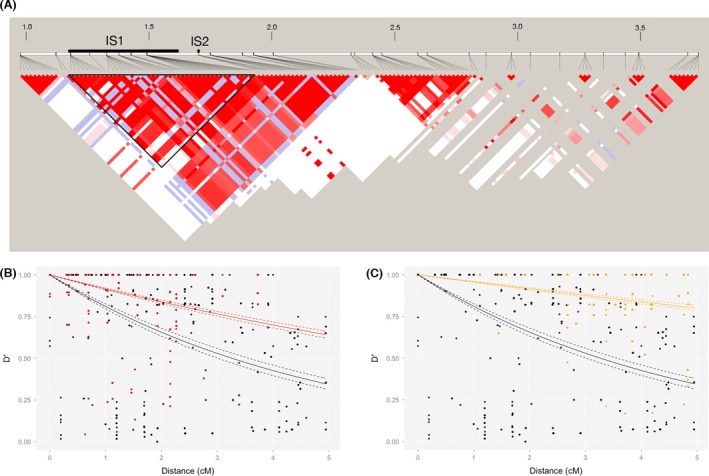
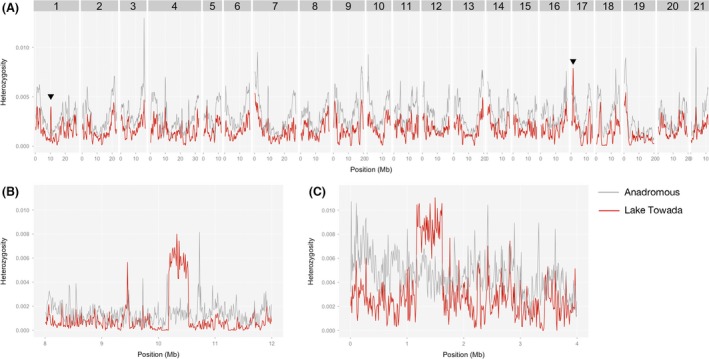
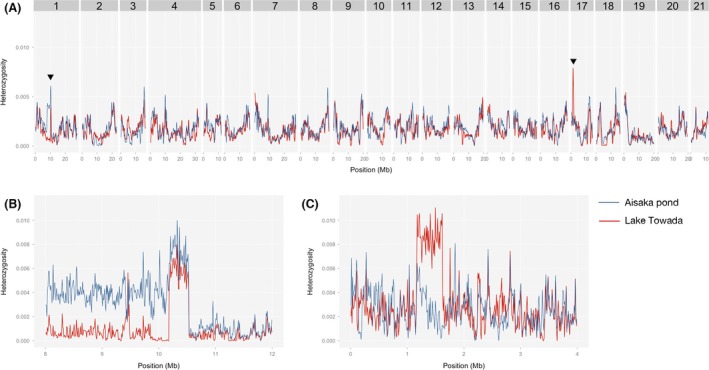
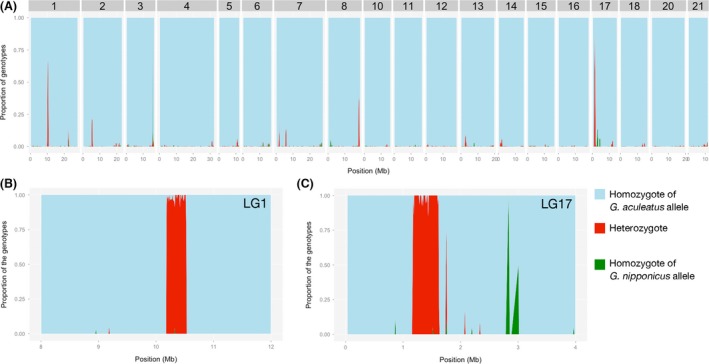
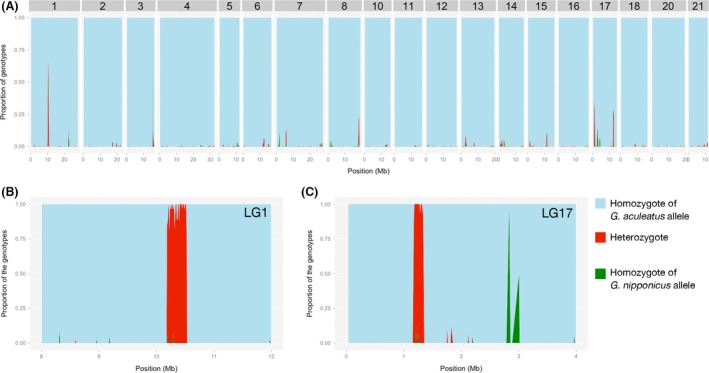
Invasive species pose a major threat to biological diversity. Although introduced populations often experience population bottlenecks, some invasive species are thought to be originated from hybridization between multiple populations or species, which can contribute to the maintenance of high genetic diversity. Recent advances in genome sequencing enable us to trace the evolutionary history of invasive species even at whole-genome level and may help to identify the history of past hybridization that may be overlooked by traditional marker-based analysis. Here, we conducted whole-genome sequencing of eight threespine stickleback (Gasterosteus aculeatus) individuals, four from a recently introduced crater lake population and four of the putative source population. We found that both populations have several small genomic regions with high genetic diversity, which resulted from introgression from a closely related species (Gasterosteus nipponicus). The sizes of the regions were too small to be detected with traditional marker-based analysis or even some reduced-representation sequencing methods. Further amplicon sequencing revealed linkage disequilibrium around an introgression site, which suggests the possibility of selective sweep at the introgression site. Thus, interspecies introgression might predate introduction and increase genetic variation in the source population. Whole-genome sequencing of even a small number of individuals can therefore provide higher resolution inference of history of introduced populations.
入侵物种对生物多样性构成重大威胁。尽管引入的种群通常会经历种群瓶颈,但一些入侵物种被认为起源于多个种群或物种之间的杂交,这可能有助于维持高遗传多样性。基因组测序的最新进展使我们能够在全基因组水平上追溯入侵物种的进化历史,并可能有助于识别过去杂交的历史,而这些历史可能会被传统的基于标记的分析所忽略。在这里,我们对8条三刺鱼(Gasterosteus aculeatus)个体进行了全基因组测序,其中4条来自最近引入的火山口湖种群,另外4条来自假定的源种群。我们发现这两个种群都有几个具有高遗传多样性的小基因组区域,这是由一个近缘物种(日本三刺鱼Gasterosteus nipponicus)的基因渗入导致的。这些区域的大小太小,无法通过传统的基于标记的分析甚至一些简化代表性测序方法检测到。进一步的扩增子测序揭示了一个基因渗入位点周围的连锁不平衡,这表明在该基因渗入位点存在选择性清除的可能性。因此,种间基因渗入可能早于引入事件,并增加了源种群中的遗传变异。因此,即使对少数个体进行全基因组测序,也可以提供对引入种群历史的更高分辨率推断。