Hilton Sarah K, Castro-Nallar Eduardo, Pérez-Losada Marcos, Toma Ian, McCaffrey Timothy A, Hoffman Eric P, Siegel Marc O, Simon Gary L, Johnson W Evan, Crandall Keith A
Computational Biology Institute, The George Washington University Ashburn, VA, USA.
Computational Biology Institute, The George Washington UniversityAshburn, VA, USA; Facultad de Ciencias Biológicas, Center for Bioinformatics and Integrative Biology, Universidad Andres BelloSantiago, Chile.
Front Microbiol. 2016 Apr 7;7:484. doi: 10.3389/fmicb.2016.00484. eCollection 2016.
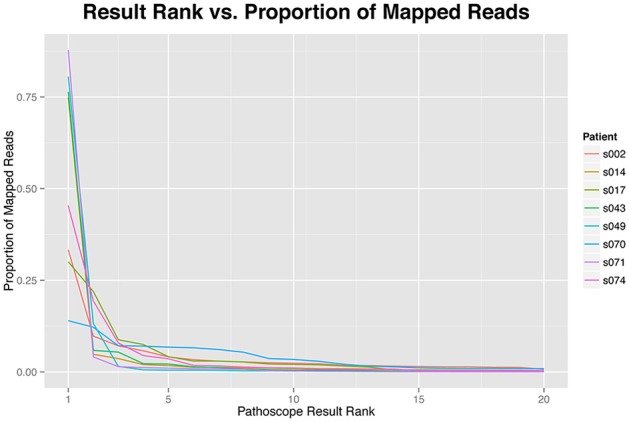
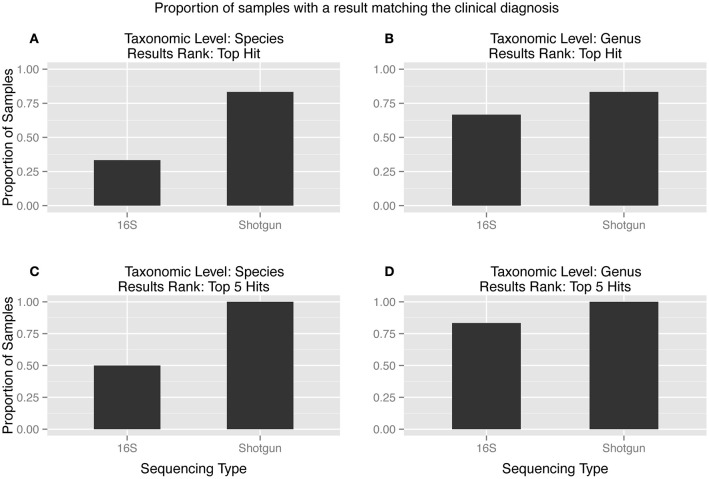
Diagnoses that are both timely and accurate are critically important for patients with life-threatening or drug resistant infections. Technological improvements in High-Throughput Sequencing (HTS) have led to its use in pathogen detection and its application in clinical diagnoses of infectious diseases. The present study compares two HTS methods, 16S rRNA marker gene sequencing (metataxonomics) and whole metagenomic shotgun sequencing (metagenomics), in their respective abilities to match the same diagnosis as traditional culture methods (culture inference) for patients with ventilator associated pneumonia (VAP). The metagenomic analysis was able to produce the same diagnosis as culture methods at the species-level for five of the six samples, while the metataxonomic analysis was only able to produce results with the same species-level identification as culture for two of the six samples. These results indicate that metagenomic analyses have the accuracy needed for a clinical diagnostic tool, but full integration in diagnostic protocols is contingent on technological improvements to decrease turnaround time and lower costs.
对于患有危及生命或耐药性感染的患者而言,及时且准确的诊断至关重要。高通量测序(HTS)技术的改进使其可用于病原体检测,并应用于传染病的临床诊断。本研究比较了两种高通量测序方法,即16S rRNA标记基因测序(宏分类学)和全宏基因组鸟枪法测序(宏基因组学),它们各自与传统培养方法(培养推断)对呼吸机相关性肺炎(VAP)患者做出相同诊断的能力。宏基因组分析能够在六个样本中的五个样本上,在物种水平上做出与培养方法相同的诊断,而宏分类学分析仅能在六个样本中的两个样本上得出与培养相同的物种水平鉴定结果。这些结果表明,宏基因组分析具备作为临床诊断工具所需的准确性,但要完全整合到诊断方案中,还取决于技术改进以缩短周转时间并降低成本。