Department of Biological Systems Engineering, Virginia Polytechnic Institute and State University, Blacksburg, Virginia, United States of America.
PLoS One. 2016 Apr 21;11(4):e0154188. doi: 10.1371/journal.pone.0154188. eCollection 2016.
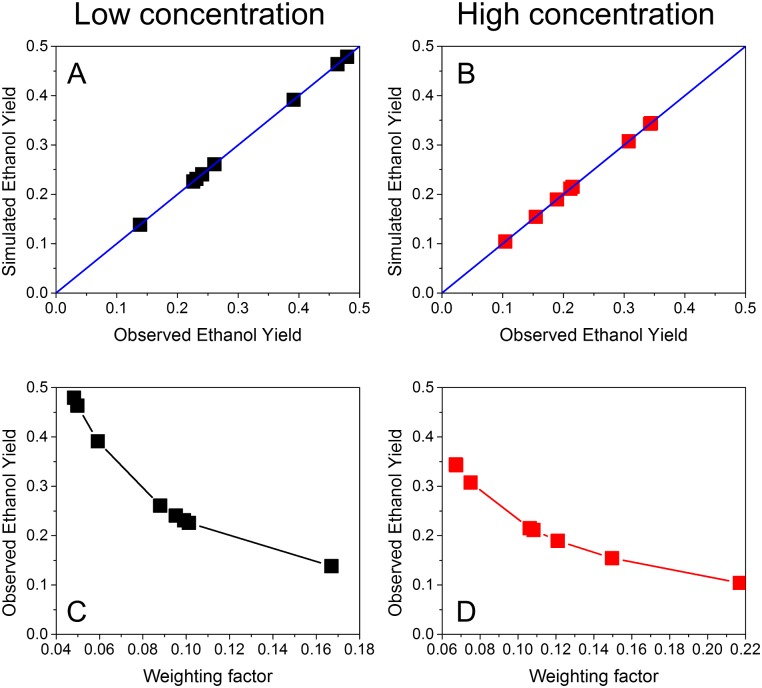
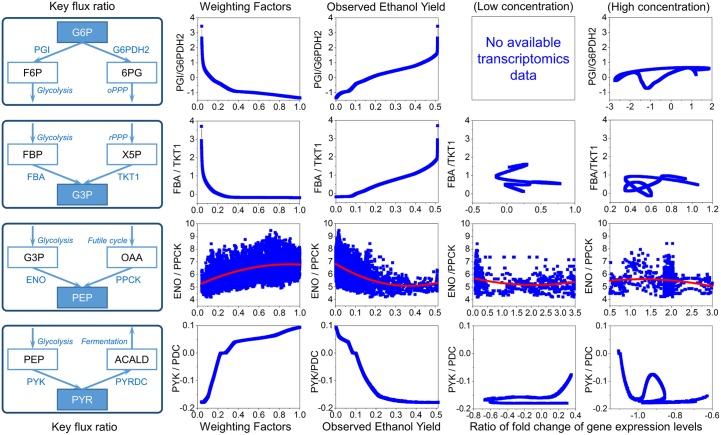
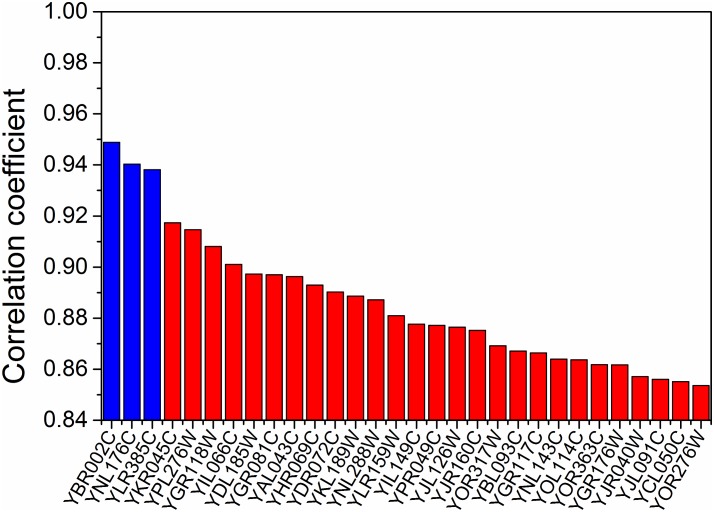
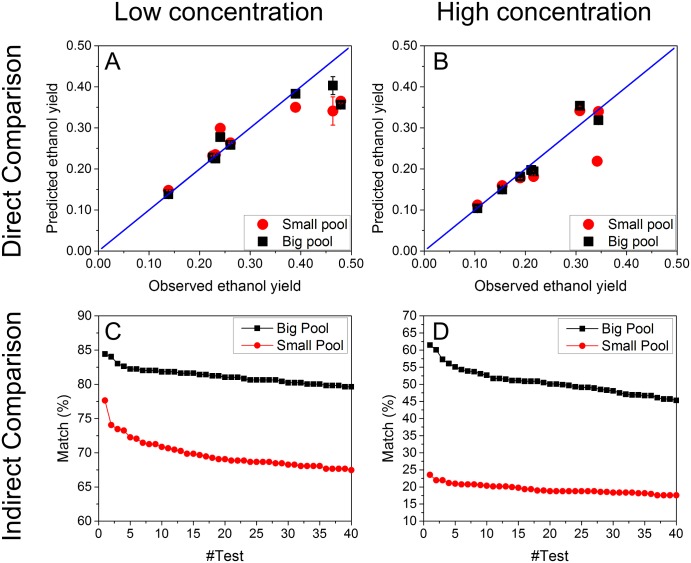
Constraint-based metabolic modeling such as flux balance analysis (FBA) has been widely used to simulate cell metabolism. Thanks to its simplicity and flexibility, numerous algorithms have been developed based on FBA and successfully predicted the phenotypes of various biological systems. However, their phenotype predictions may not always be accurate in FBA because of using the objective function that is assumed for cell metabolism. To overcome this challenge, we have developed a novel computational framework, namely omFBA, to integrate multi-omics data (e.g. transcriptomics) into FBA to obtain omics-guided objective functions with high accuracy. In general, we first collected transcriptomics data and phenotype data from published database (e.g. GEO database) for different microorganisms such as Saccharomyces cerevisiae. We then developed a "Phenotype Match" algorithm to derive an objective function for FBA that could lead to the most accurate estimation of the known phenotype (e.g. ethanol yield). The derived objective function was next correlated with the transcriptomics data via regression analysis to generate the omics-guided objective function, which was next used to accurately simulate cell metabolism at unknown conditions. We have applied omFBA in studying sugar metabolism of S. cerevisiae and found that the ethanol yield could be accurately predicted in most of the cases tested (>80%) by using transcriptomics data alone, and revealed valuable metabolic insights such as the dynamics of flux ratios. Overall, omFBA presents a novel platform to potentially integrate multi-omics data simultaneously and could be incorporated with other FBA-derived tools by replacing the arbitrary objective function with the omics-guided objective functions.
基于约束的代谢建模,如通量平衡分析(FBA),已被广泛用于模拟细胞代谢。由于其简单性和灵活性,基于 FBA 已经开发了许多算法,并成功预测了各种生物系统的表型。然而,由于使用了假设的细胞代谢目标函数,它们的表型预测在 FBA 中并不总是准确的。为了克服这一挑战,我们开发了一种新的计算框架,即 omFBA,将多组学数据(如转录组学)整合到 FBA 中,以获得具有高精度的组学指导目标函数。通常,我们首先从不同的微生物(如酿酒酵母)的已发表数据库(如 GEO 数据库)中收集转录组学数据和表型数据。然后,我们开发了一种“表型匹配”算法,为 FBA 导出一个目标函数,该函数可以对已知表型(例如乙醇产量)进行最准确的估计。接下来,通过回归分析将导出的目标函数与转录组学数据相关联,生成组学指导的目标函数,然后使用该函数在未知条件下准确模拟细胞代谢。我们已经将 omFBA 应用于研究酿酒酵母的糖代谢,发现仅使用转录组学数据就可以在大多数测试案例中(>80%)准确预测乙醇产量,并揭示了有价值的代谢见解,如通量比的动态。总体而言,omFBA 提供了一个新的平台,可以同时潜在地整合多组学数据,并通过用组学指导的目标函数替换任意目标函数,与其他基于 FBA 的工具相结合。