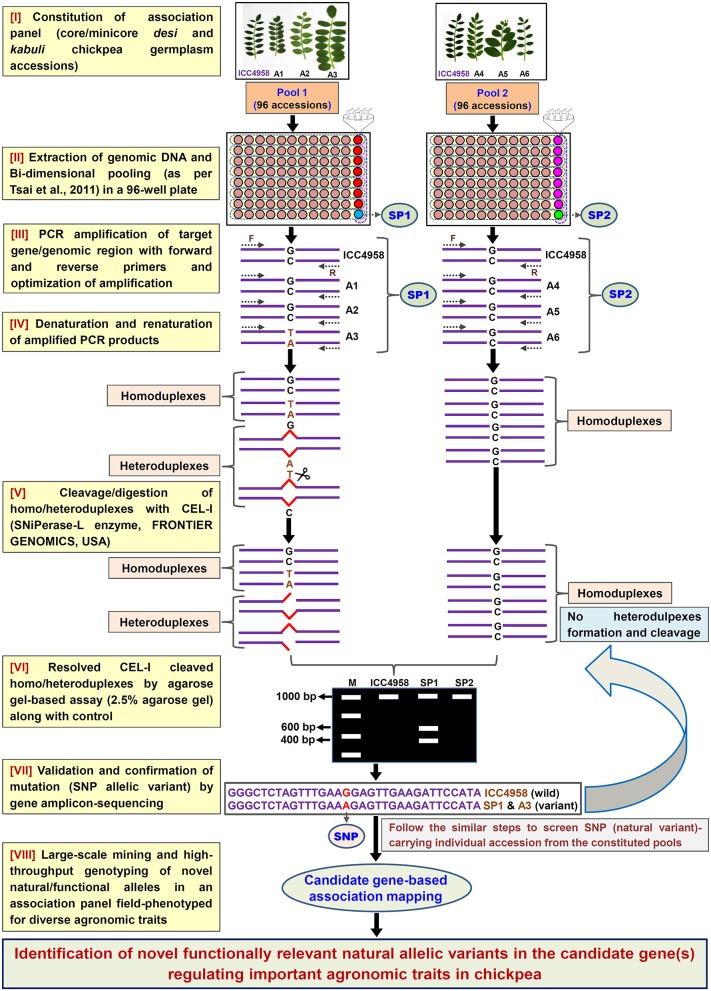
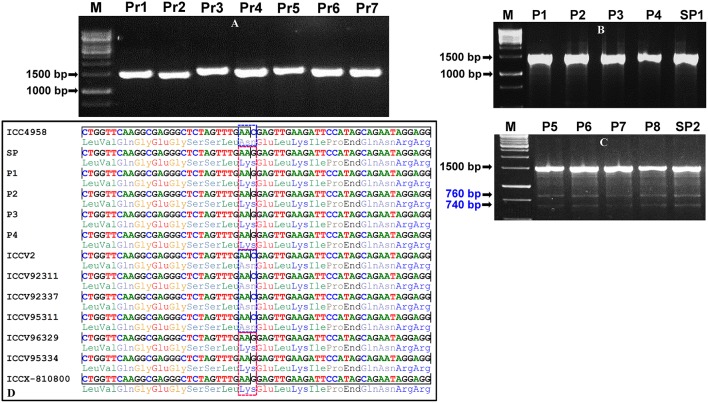
Bajaj Deepak, Srivastava Rishi, Nath Manoj, Tripathi Shailesh, Bharadwaj Chellapilla, Upadhyaya Hari D, Tyagi Akhilesh K, Parida Swarup K
Govt. of India, Plant Genomics and Molecular Breeding Lab, Department of Biotechnology, National Institute of Plant Genome Research New Delhi, India.
National Research Centre on Plant Biotechnology New Delhi, India.
Front Plant Sci. 2016 Apr 19;7:450. doi: 10.3389/fpls.2016.00450. eCollection 2016.
The large-scale mining and high-throughput genotyping of novel gene-based allelic variants in natural mapping population are essential for association mapping to identify functionally relevant molecular tags governing useful agronomic traits in chickpea. The present study employs an alternative time-saving, non-laborious and economical pool-based EcoTILLING approach coupled with agarose gel detection assay to discover 1133 novel SNP allelic variants from diverse coding and regulatory sequence components of 1133 transcription factor (TF) genes by genotyping in 192 diverse desi and kabuli chickpea accessions constituting a seed weight association panel. Integrating these SNP genotyping data with seed weight field phenotypic information of 192 structured association panel identified eight SNP alleles in the eight TF genes regulating seed weight of chickpea. The associated individual and combination of all SNPs explained 10-15 and 31% phenotypic variation for seed weight, respectively. The EcoTILLING-based large-scale allele mining and genotyping strategy implemented for association mapping is found much effective for a diploid genome crop species like chickpea with narrow genetic base and low genetic polymorphism. This optimized approach thus can be deployed for various genomics-assisted breeding applications with optimal expense of resources in domesticated chickpea. The seed weight-associated natural allelic variants and candidate TF genes delineated have potential to accelerate marker-assisted genetic improvement of chickpea.
在天然作图群体中对基于基因的新型等位变异进行大规模挖掘和高通量基因分型,对于关联作图以鉴定控制鹰嘴豆有用农艺性状的功能相关分子标记至关重要。本研究采用一种省时、省力且经济的基于混合样本的EcoTILLING方法,并结合琼脂糖凝胶检测分析,通过对构成种子重量关联群体的192份不同的鹰嘴豆品种(包括本地鹰嘴豆和卡布利鹰嘴豆)进行基因分型,从1133个转录因子(TF)基因的不同编码和调控序列元件中发现了1133个新型SNP等位变异。将这些SNP基因分型数据与192份结构化关联群体的种子重量田间表型信息相结合,鉴定出调控鹰嘴豆种子重量的8个TF基因中的8个SNP等位基因。所有SNP的相关个体及组合分别解释了种子重量10%-15%和31%的表型变异。基于EcoTILLING的大规模等位基因挖掘和基因分型策略在关联作图中的应用,对于像鹰嘴豆这样遗传基础狭窄且遗传多态性低的二倍体基因组作物物种非常有效。因此,这种优化方法可用于鹰嘴豆的各种基因组辅助育种应用,并以最优的资源消耗实现。所描绘的与种子重量相关的天然等位变异和候选TF基因有潜力加速鹰嘴豆的标记辅助遗传改良。