Bolleman Jerven T, Mungall Christopher J, Strozzi Francesco, Baran Joachim, Dumontier Michel, Bonnal Raoul J P, Buels Robert, Hoehndorf Robert, Fujisawa Takatomo, Katayama Toshiaki, Cock Peter J A
Swiss-Prot group, SIB Swiss Institute of Bioinformatics, Centre Medical Universitaire, 1 rue Michel, Servet, Geneva 4, 1211, Switzerland.
Genomics Division, Lawrence Berkeley National Laboratory, Berkeley, 94720, CA, US.
J Biomed Semantics. 2016 Jun 13;7:39. doi: 10.1186/s13326-016-0067-z.
Nucleotide and protein sequence feature annotations are essential to understand biology on the genomic, transcriptomic, and proteomic level. Using Semantic Web technologies to query biological annotations, there was no standard that described this potentially complex location information as subject-predicate-object triples.
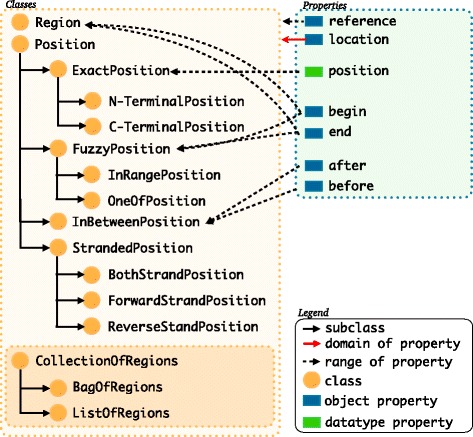
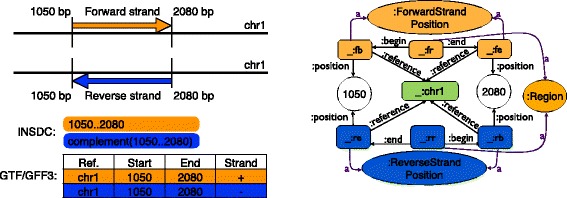
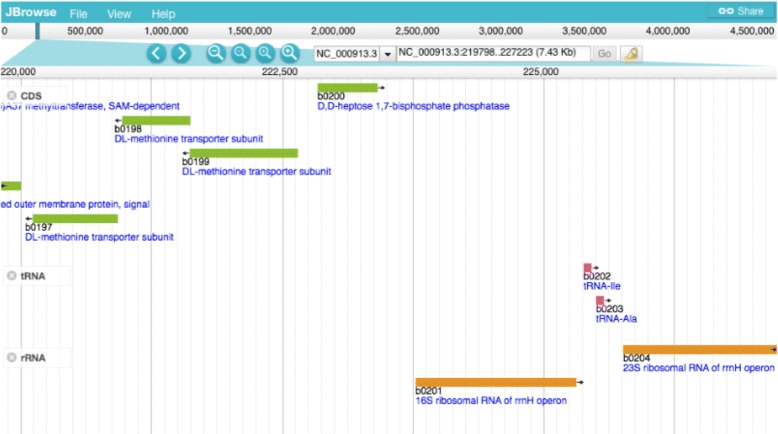
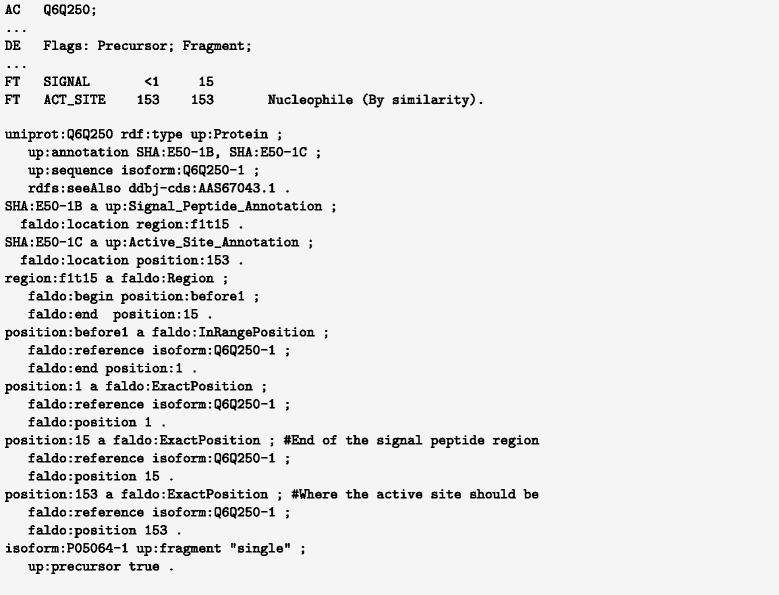
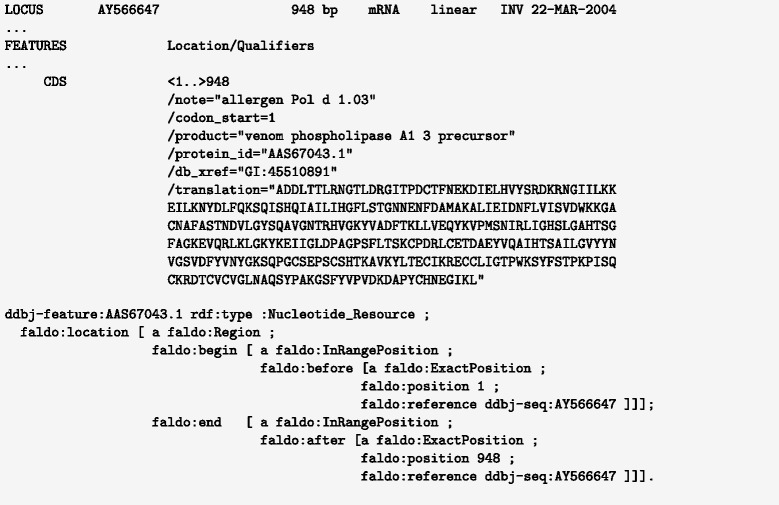
We have developed an ontology, the Feature Annotation Location Description Ontology (FALDO), to describe the positions of annotated features on linear and circular sequences. FALDO can be used to describe nucleotide features in sequence records, protein annotations, and glycan binding sites, among other features in coordinate systems of the aforementioned "omics" areas. Using the same data format to represent sequence positions that are independent of file formats allows us to integrate sequence data from multiple sources and data types. The genome browser JBrowse is used to demonstrate accessing multiple SPARQL endpoints to display genomic feature annotations, as well as protein annotations from UniProt mapped to genomic locations.
Our ontology allows users to uniformly describe - and potentially merge - sequence annotations from multiple sources. Data sources using FALDO can prospectively be retrieved using federalised SPARQL queries against public SPARQL endpoints and/or local private triple stores.
核苷酸和蛋白质序列特征注释对于在基因组、转录组和蛋白质组水平上理解生物学至关重要。使用语义网技术查询生物学注释时,没有标准将这种潜在复杂的位置信息描述为主谓宾三元组。
我们开发了一种本体,即特征注释位置描述本体(FALDO),用于描述线性和环状序列上注释特征的位置。FALDO可用于描述序列记录中的核苷酸特征、蛋白质注释和聚糖结合位点,以及上述“组学”领域坐标系中的其他特征。使用相同的数据格式来表示与文件格式无关的序列位置,使我们能够整合来自多个来源和数据类型的序列数据。基因组浏览器JBrowse用于演示如何访问多个SPARQL端点以显示基因组特征注释,以及映射到基因组位置的来自UniProt的蛋白质注释。
我们的本体允许用户统一描述并可能合并来自多个来源的序列注释。使用FALDO的数据源可以通过针对公共SPARQL端点和/或本地私有三元组存储的联邦化SPARQL查询进行前瞻性检索。