Bruder Katherine, Malki Kema, Cooper Alexandria, Sible Emily, Shapiro Jason W, Watkins Siobhan C, Putonti Catherine
Department of Biology, Loyola University Chicago, Chicago, IL, USA.
Department of Biology, Loyola University Chicago, Chicago, IL, USA.; Bioinformatics Program, Loyola University Chicago, Chicago, IL, USA.
Evol Bioinform Online. 2016 Jun 20;12(Suppl 1):25-33. doi: 10.4137/EBO.S38549. eCollection 2016.
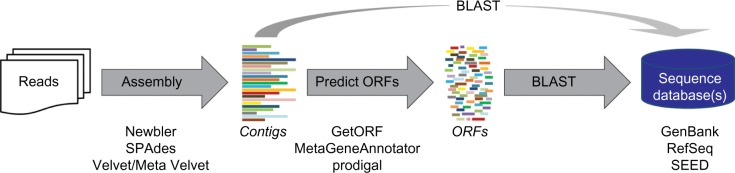
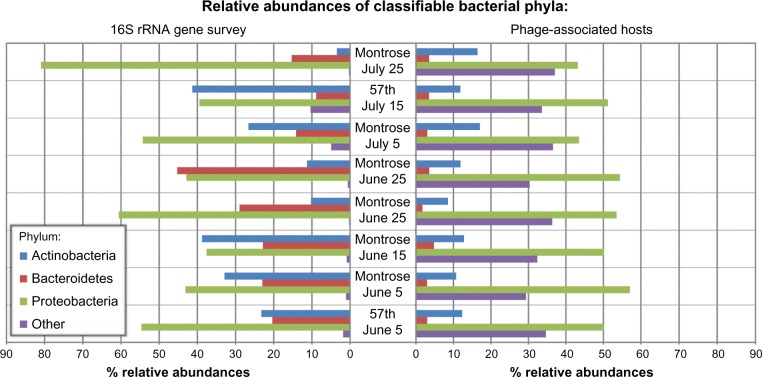
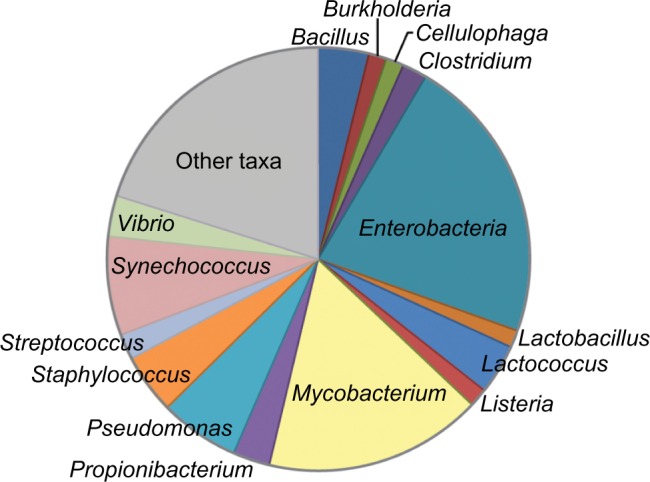
Advances in bioinformatics and sequencing technologies have allowed for the analysis of complex microbial communities at an unprecedented rate. While much focus is often placed on the cellular members of these communities, viruses play a pivotal role, particularly bacteria-infecting viruses (bacteriophages); phages mediate global biogeochemical processes and drive microbial evolution through bacterial grazing and horizontal gene transfer. Despite their importance and ubiquity in nature, very little is known about the diversity and structure of viral communities. Though the need for culture-based methods for viral identification has been somewhat circumvented through metagenomic techniques, the analysis of metaviromic data is marred with many unique issues. In this review, we examine the current bioinformatic approaches for metavirome analyses and the inherent challenges facing the field as illustrated by the ongoing efforts in the exploration of freshwater phage populations.
生物信息学和测序技术的进步使得以前所未有的速度分析复杂的微生物群落成为可能。虽然人们常常将大量注意力放在这些群落的细胞成员上,但病毒发挥着关键作用,尤其是感染细菌的病毒(噬菌体);噬菌体介导全球生物地球化学过程,并通过细菌捕食和水平基因转移推动微生物进化。尽管它们在自然界中具有重要性和普遍性,但人们对病毒群落的多样性和结构却知之甚少。虽然通过宏基因组技术在一定程度上规避了基于培养的病毒鉴定方法的需求,但宏病毒组数据分析却存在许多独特问题。在本综述中,我们研究了当前用于宏病毒组分析的生物信息学方法,以及通过对淡水噬菌体群体的探索所展现出的该领域面临的固有挑战。