Garretto Andrea, Hatzopoulos Thomas, Putonti Catherine
Bioinformatics Program, Loyola University of Chicago, Chicago, IL, United States of America.
Department of Computer Science, Loyola University of Chicago, Chicago, IL, United States of America.
PeerJ. 2019 Apr 10;7:e6695. doi: 10.7717/peerj.6695. eCollection 2019.
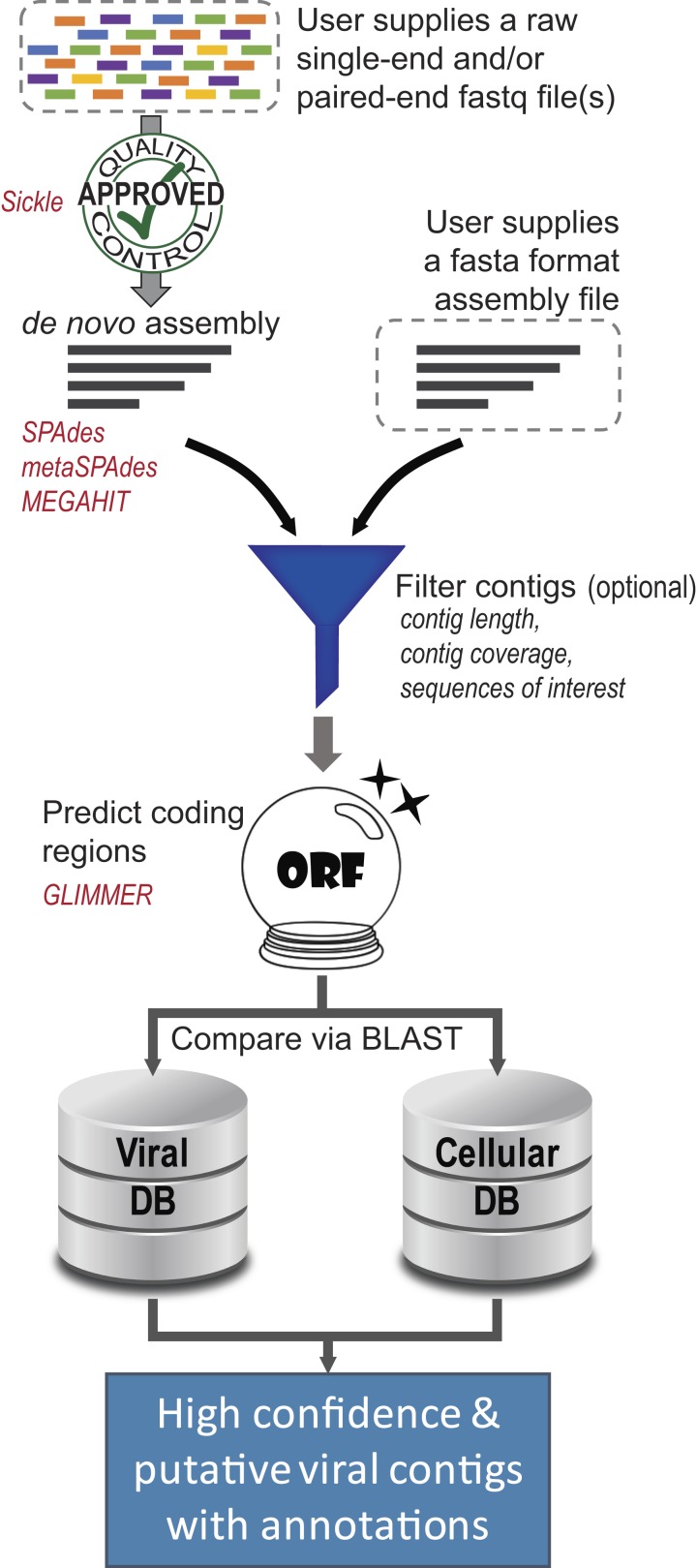
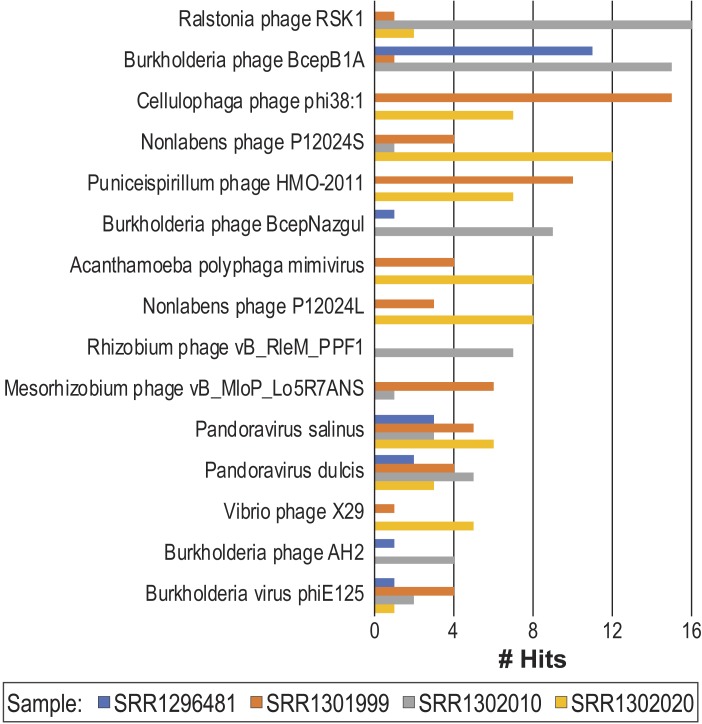
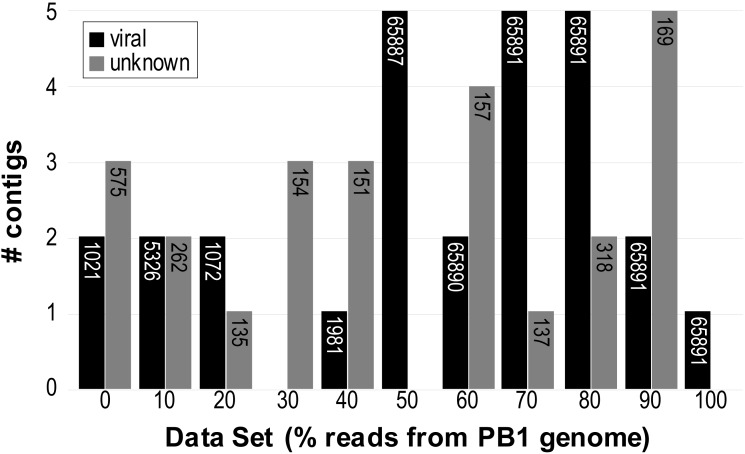
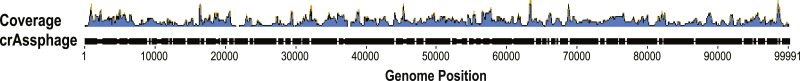
Metagenomics has enabled sequencing of viral communities from a myriad of different environments. Viral metagenomic studies routinely uncover sequences with no recognizable homology to known coding regions or genomes. Nevertheless, complete viral genomes have been constructed directly from complex community metagenomes, often through tedious manual curation. To address this, we developed the software tool virMine to identify viral genomes from raw reads representative of viral or mixed (viral and bacterial) communities. virMine automates sequence read quality control, assembly, and annotation. Researchers can easily refine their search for a specific study system and/or feature(s) of interest. In contrast to other viral genome detection tools that often rely on the recognition of viral signature sequences, virMine is not restricted by the insufficient representation of viral diversity in public data repositories. Rather, viral genomes are identified through an iterative approach, first omitting non-viral sequences. Thus, both relatives of previously characterized viruses and novel species can be detected, including both eukaryotic viruses and bacteriophages. Here we present virMine and its analysis of synthetic communities as well as metagenomic data sets from three distinctly different environments: the gut microbiota, the urinary microbiota, and freshwater viromes. Several new viral genomes were identified and annotated, thus contributing to our understanding of viral genetic diversity in these three environments.
宏基因组学已能够对来自无数不同环境的病毒群落进行测序。病毒宏基因组学研究经常发现与已知编码区或基因组没有可识别同源性的序列。然而,完整的病毒基因组通常是通过繁琐的人工整理直接从复杂的群落宏基因组构建而来的。为了解决这个问题,我们开发了软件工具virMine,用于从代表病毒或混合(病毒和细菌)群落的原始 reads 中识别病毒基因组。virMine 实现了序列读取质量控制、组装和注释的自动化。研究人员可以轻松地针对特定的研究系统和/或感兴趣的特征优化搜索。与其他通常依赖于识别病毒特征序列的病毒基因组检测工具不同,virMine 不受公共数据存储库中病毒多样性代表性不足的限制。相反,病毒基因组是通过一种迭代方法识别的,首先去除非病毒序列。因此,既可以检测到先前已表征病毒的亲属,也可以检测到新物种,包括真核病毒和噬菌体。在这里,我们展示了 virMine 及其对合成群落以及来自三种截然不同环境的宏基因组数据集的分析:肠道微生物群、泌尿微生物群和淡水病毒组。识别并注释了几个新的病毒基因组,从而有助于我们了解这三种环境中的病毒遗传多样性。