Laffy Patrick W, Wood-Charlson Elisha M, Turaev Dmitrij, Weynberg Karen D, Botté Emmanuelle S, van Oppen Madeleine J H, Webster Nicole S, Rattei Thomas
Australian Institute of Marine Science Townsville, QLD, Australia.
Center for Microbial Oceanography: Research and Education, University of Hawai'i at Mānoa Honolulu, HI, USA.
Front Microbiol. 2016 Jun 9;7:822. doi: 10.3389/fmicb.2016.00822. eCollection 2016.
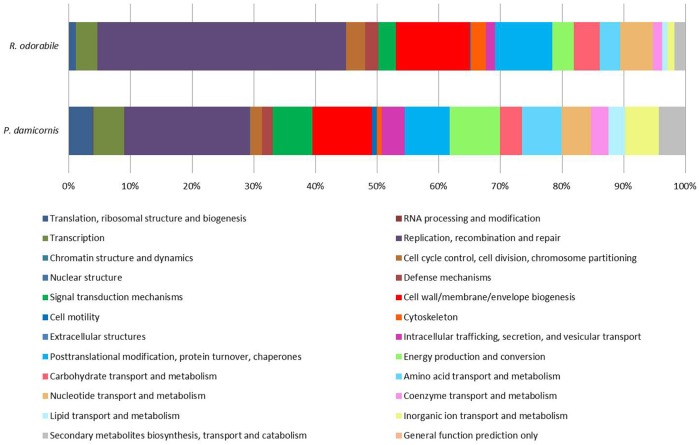
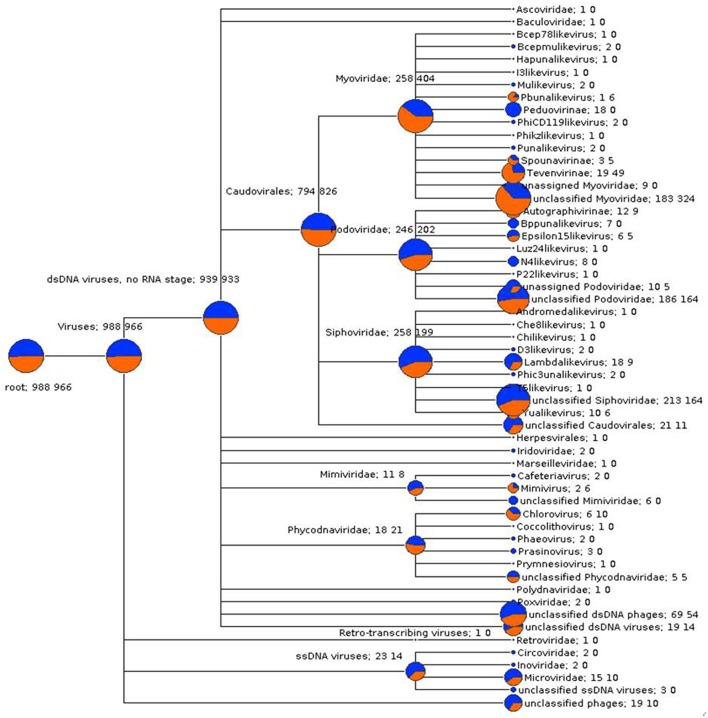
Abundant bioinformatics resources are available for the study of complex microbial metagenomes, however their utility in viral metagenomics is limited. HoloVir is a robust and flexible data analysis pipeline that provides an optimized and validated workflow for taxonomic and functional characterization of viral metagenomes derived from invertebrate holobionts. Simulated viral metagenomes comprising varying levels of viral diversity and abundance were used to determine the optimal assembly and gene prediction strategy, and multiple sequence assembly methods and gene prediction tools were tested in order to optimize our analysis workflow. HoloVir performs pairwise comparisons of single read and predicted gene datasets against the viral RefSeq database to assign taxonomy and additional comparison to phage-specific and cellular markers is undertaken to support the taxonomic assignments and identify potential cellular contamination. Broad functional classification of the predicted genes is provided by assignment of COG microbial functional category classifications using EggNOG and higher resolution functional analysis is achieved by searching for enrichment of specific Swiss-Prot keywords within the viral metagenome. Application of HoloVir to viral metagenomes from the coral Pocillopora damicornis and the sponge Rhopaloeides odorabile demonstrated that HoloVir provides a valuable tool to characterize holobiont viral communities across species, environments, or experiments.
有丰富的生物信息学资源可用于复杂微生物宏基因组的研究,然而它们在病毒宏基因组学中的效用有限。HoloVir是一个强大且灵活的数据分析流程,为源自无脊椎动物全生物的病毒宏基因组的分类和功能表征提供了优化和经过验证的工作流程。使用包含不同水平病毒多样性和丰度的模拟病毒宏基因组来确定最佳组装和基因预测策略,并测试了多种序列组装方法和基因预测工具,以优化我们的分析工作流程。HoloVir将单读段和预测基因数据集与病毒RefSeq数据库进行成对比较以进行分类,并且还会与噬菌体特异性和细胞标记进行额外比较,以支持分类任务并识别潜在的细胞污染。通过使用EggNOG分配COG微生物功能类别分类来提供预测基因的广泛功能分类,并通过在病毒宏基因组中搜索特定的Swiss-Prot关键词的富集来实现更高分辨率的功能分析。将HoloVir应用于来自鹿角杯形珊瑚和异味圆海绵的病毒宏基因组表明,HoloVir为跨物种、环境或实验表征全生物病毒群落提供了一个有价值的工具。