Lim Jaechul, Lee Mihye, Son Ahyeon, Chang Hyeshik, Kim V Narry
Center for RNA Research, Institute for Basic Science, Seoul 08826, Korea; School of Biological Sciences, Seoul National University, Seoul 08826, Korea.
Genes Dev. 2016 Jul 15;30(14):1671-82. doi: 10.1101/gad.284802.116. Epub 2016 Jul 21.
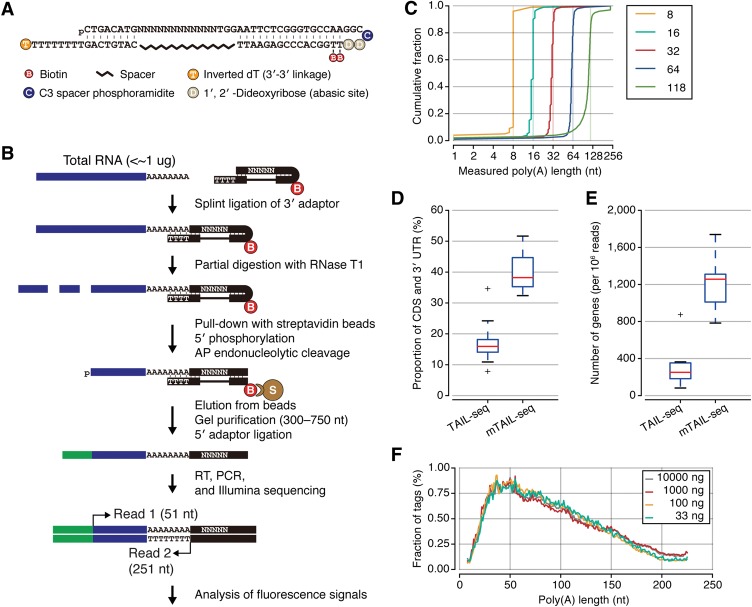
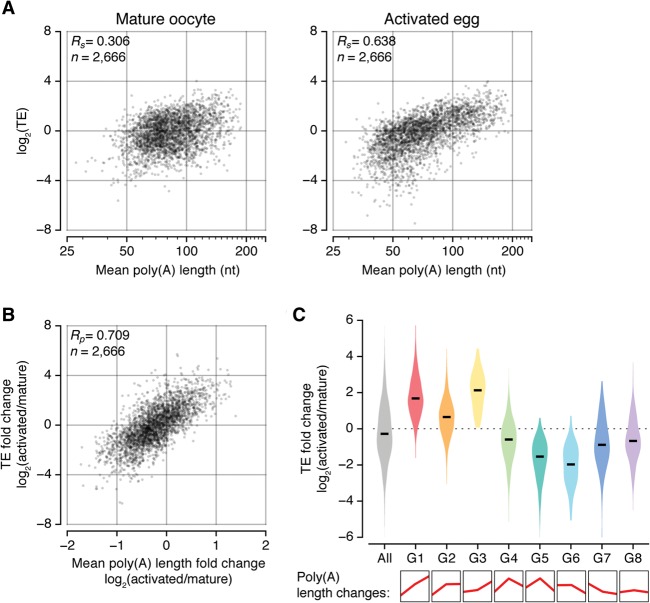
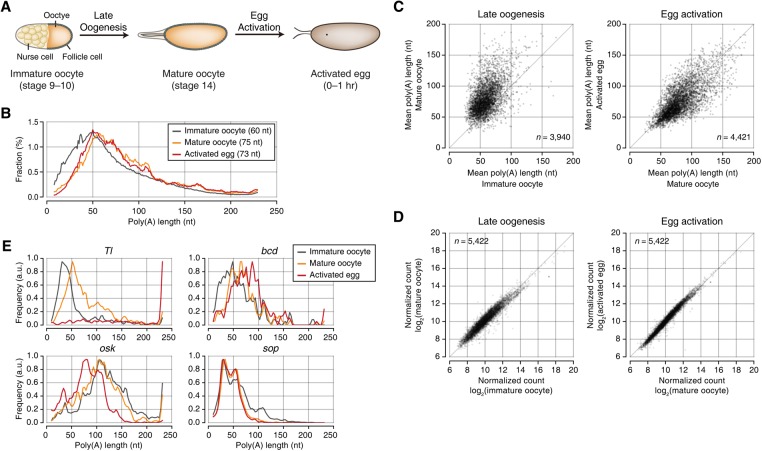
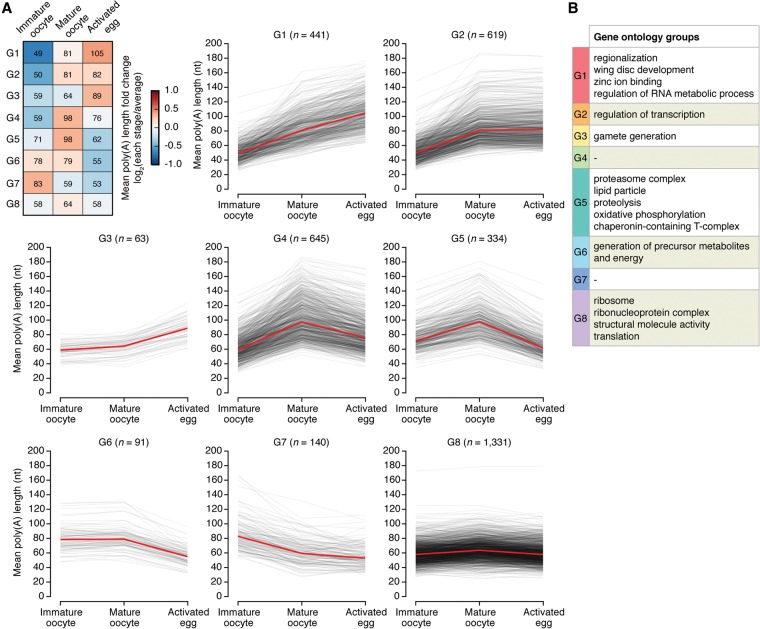
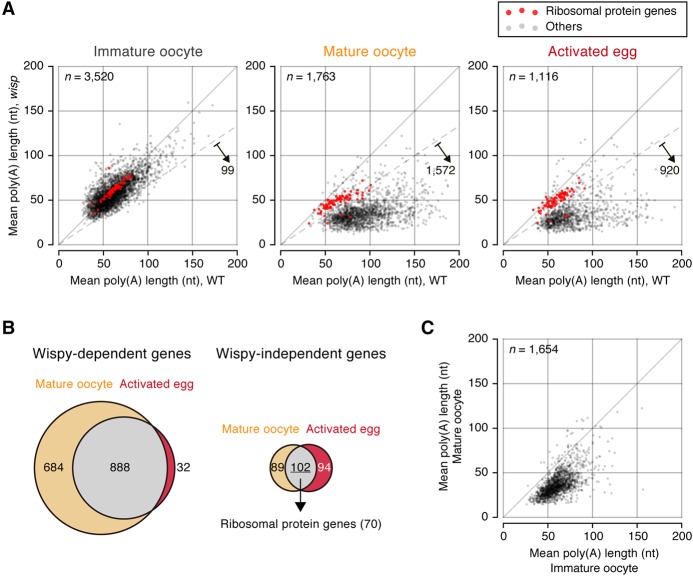
Eukaryotic mRNAs are subject to multiple types of tailing that critically influence mRNA stability and translatability. To investigate RNA tails at the genomic scale, we previously developed TAIL-seq, but its low sensitivity precluded its application to biological materials of minute quantity. In this study, we report a new version of TAIL-seq (mRNA TAIL-seq [mTAIL-seq]) with enhanced sequencing depth for mRNAs (by ∼1000-fold compared with the previous version). The improved method allows us to investigate the regulation of poly(A) tails in Drosophila oocytes and embryos. We found that maternal mRNAs are polyadenylated mainly during late oogenesis, prior to fertilization, and that further modulation occurs upon egg activation. Wispy, a noncanonical poly(A) polymerase, adenylates the vast majority of maternal mRNAs, with a few intriguing exceptions such as ribosomal protein transcripts. By comparing mTAIL-seq data with ribosome profiling data, we found a strong coupling between poly(A) tail length and translational efficiency during egg activation. Our data suggest that regulation of poly(A) tails in oocytes shapes the translatomic landscape of embryos, thereby directing the onset of animal development. By virtue of the high sensitivity, low cost, technical robustness, and broad accessibility, mTAIL-seq will be a potent tool to improve our understanding of mRNA tailing in diverse biological systems.
真核生物的信使核糖核酸(mRNA)会经历多种类型的加尾过程,这些过程对mRNA的稳定性和可翻译性有着至关重要的影响。为了在基因组规模上研究RNA尾巴,我们之前开发了TAIL-seq,但它的低灵敏度使其无法应用于微量生物材料。在本研究中,我们报告了一种新版本的TAIL-seq(mRNA TAIL-seq [mTAIL-seq]),其对mRNA的测序深度有所增强(与之前版本相比提高了约1000倍)。这种改进后的方法使我们能够研究果蝇卵母细胞和胚胎中聚腺苷酸(poly(A))尾巴的调控情况。我们发现,母源mRNA主要在卵子发生后期、受精之前进行多聚腺苷酸化,并且在卵子激活时会发生进一步的调控。Wispy是一种非典型的多聚(A)聚合酶,它对绝大多数母源mRNA进行腺苷酸化,只有少数有趣的例外情况,如核糖体蛋白转录本。通过将mTAIL-seq数据与核糖体谱分析数据进行比较,我们发现在卵子激活过程中,poly(A)尾巴长度与翻译效率之间存在很强的耦合关系。我们的数据表明,卵母细胞中poly(A)尾巴的调控塑造了胚胎的翻译组景观,从而指导动物发育的开始。凭借高灵敏度、低成本、技术稳健性和广泛的可及性,mTAIL-seq将成为一种有力的工具,有助于我们更好地理解不同生物系统中的mRNA加尾情况。