Salvetti Elisa, Campanaro Stefano, Campedelli Ilenia, Fracchetti Fabio, Gobbi Alex, Tornielli Giovanni Battista, Torriani Sandra, Felis Giovanna E
Department of Biotechnology, University of Verona Verona, Italy.
Department of Biology, University of Padova Padua, Italy.
Front Microbiol. 2016 Jun 23;7:937. doi: 10.3389/fmicb.2016.00937. eCollection 2016.
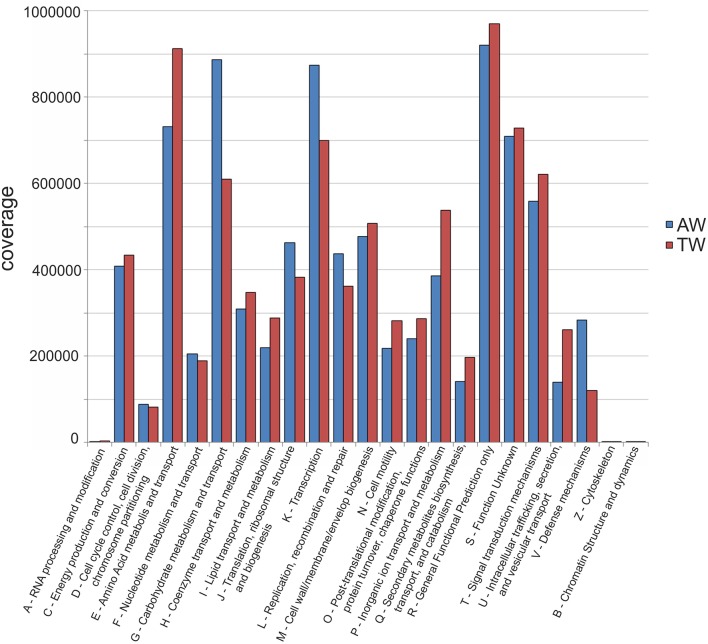
Vitis vinifera L. cv. Corvina grape forms the basis for the production of unique wines, such as Amarone, whose distinctive sensory features are strongly linked to the post-harvest grape withering process. Indeed, this process increases sugar concentration and changes must characteristics. While microorganisms involved in must fermentation have been widely investigated, few data are available on the microbiota of withered grapes. Thus, in this paper, a whole metagenome sequencing (WMS) approach was used to analyse the microbial consortium associated with Corvina berries at the end of the withering process performed in two different conditions ("traditional withering," TW or "accelerated withering," AW), and to unveil whether changes of drying parameters could have an impact on microbial diversity. Samples of healthy undamaged berries were collected and washed, to recover microorganisms from the surface and avoid contamination with grapevine genetic material. Isolated DNA was sequenced and the data obtained were analyzed with several bioinformatics methods. The eukaryotic community was mainly composed by members of the phylum Ascomycota, including Eurotiomycetes, Sordariomycetes, and Dothideomycetes. Moreover, the distribution of the genera Aspergillus and Penicillium (class Eurotiomycetes) varied between the withered berry samples. Instead, Botryotinia, Saccharomyces, and other wine technologically useful microorganisms were relatively scarce in both samples. For prokaryotes, 25 phyla were identified, nine of which were common to both conditions. Environmental bacteria belonging to the class Gammaproteobacteria were dominant and, in particular, the TW sample was characterized by members of the family Pseudomonadaceae, while members of the family Enterobacteriaceae dominated the AW sample, in addition to Sphyngobacteria and Clostridia. Finally, the binning procedure discovered 15 putative genomes which dominated the microbial community of the two samples, and included representatives of genera Erwinia, Pantoea, Pseudomonas, Clostridium, Paenibacillus, and of orders Lactobacillales and Actinomycetales. These results provide insights into the microbial consortium of Corvina withered berries and reveal relevant variations attributable to post-harvest withering conditions, underling how WMS could open novel perspectives in the knowledge and management of the withering process of Corvina, with an impact on the winemaking of important Italian wines.
科维纳(Corvina)葡萄是酿酒葡萄品种,是生产独特葡萄酒(如阿玛罗尼葡萄酒)的基础,其独特的感官特征与收获后葡萄的风干过程密切相关。事实上,这个过程会提高糖分浓度并改变葡萄汁的特性。虽然参与葡萄汁发酵的微生物已得到广泛研究,但关于风干葡萄微生物群的数据却很少。因此,本文采用全基因组测序(WMS)方法,分析在两种不同条件下(“传统风干”,TW 或“加速风干”,AW)进行的风干过程结束时与科维纳浆果相关的微生物群落,并揭示干燥参数的变化是否会对微生物多样性产生影响。收集健康无损的浆果样本并进行清洗,以从表面回收微生物并避免葡萄遗传物质的污染。对分离出的 DNA 进行测序,并使用多种生物信息学方法对获得的数据进行分析。真核生物群落主要由子囊菌门的成员组成,包括散囊菌纲、粪壳菌纲和座囊菌纲。此外,曲霉菌属和青霉菌属(散囊菌纲)的分布在风干浆果样本之间有所不同。相反,葡萄孢属、酿酒酵母属和其他对葡萄酒酿造有用的微生物在两个样本中相对较少。对于原核生物,鉴定出 25 个门,其中 9 个门在两种条件下都很常见。属于γ-变形菌纲的环境细菌占主导地位,特别是 TW 样本的特征是假单胞菌科的成员,而肠杆菌科的成员在 AW 样本中占主导地位,此外还有鞘脂杆菌纲和梭菌纲。最后,分箱程序发现了 15 个假定基因组,它们主导了两个样本的微生物群落,包括欧文氏菌属、泛菌属、假单胞菌属、梭菌属、芽孢杆菌属的代表,以及乳杆菌目和放线菌目的代表。这些结果为科维纳风干浆果的微生物群落提供了见解,并揭示了收获后风干条件导致的相关变化,强调了 WMS 如何能为科维纳风干过程的知识和管理开辟新的视角,对重要意大利葡萄酒的酿造产生影响。