Lim Chee Yee, Wang Huange, Woodhouse Steven, Piterman Nir, Wernisch Lorenz, Fisher Jasmin, Göttgens Berthold
Department of Haematology, Wellcome Trust and MRC Cambridge Stem Cell Institute, Cambridge Institute for Medical Research, University of Cambridge, Hills Road, Cambridge, CB2 0XY, UK.
Department of Computer Science, University of Leicester, Leicester, UK.
BMC Bioinformatics. 2016 Sep 6;17(1):355. doi: 10.1186/s12859-016-1235-y.
Rapid technological innovation for the generation of single-cell genomics data presents new challenges and opportunities for bioinformatics analysis. One such area lies in the development of new ways to train gene regulatory networks. The use of single-cell expression profiling technique allows the profiling of the expression states of hundreds of cells, but these expression states are typically noisier due to the presence of technical artefacts such as drop-outs. While many algorithms exist to infer a gene regulatory network, very few of them are able to harness the extra expression states present in single-cell expression data without getting adversely affected by the substantial technical noise present.
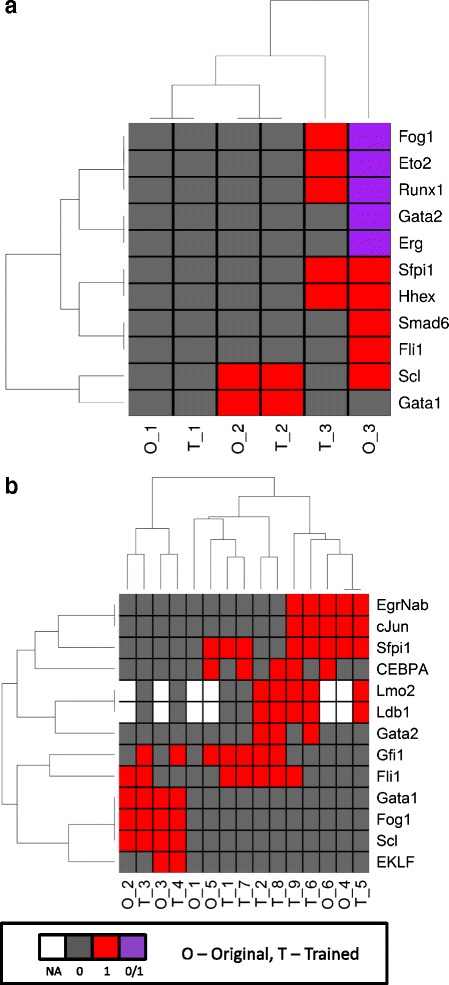
Here we introduce BTR, an algorithm for training asynchronous Boolean models with single-cell expression data using a novel Boolean state space scoring function. BTR is capable of refining existing Boolean models and reconstructing new Boolean models by improving the match between model prediction and expression data. We demonstrate that the Boolean scoring function performed favourably against the BIC scoring function for Bayesian networks. In addition, we show that BTR outperforms many other network inference algorithms in both bulk and single-cell synthetic expression data. Lastly, we introduce two case studies, in which we use BTR to improve published Boolean models in order to generate potentially new biological insights.
BTR provides a novel way to refine or reconstruct Boolean models using single-cell expression data. Boolean model is particularly useful for network reconstruction using single-cell data because it is more robust to the effect of drop-outs. In addition, BTR does not assume any relationship in the expression states among cells, it is useful for reconstructing a gene regulatory network with as few assumptions as possible. Given the simplicity of Boolean models and the rapid adoption of single-cell genomics by biologists, BTR has the potential to make an impact across many fields of biomedical research.
单细胞基因组学数据生成方面的快速技术创新给生物信息学分析带来了新的挑战和机遇。其中一个领域在于开发训练基因调控网络的新方法。单细胞表达谱技术的应用使得能够对数百个细胞的表达状态进行分析,但由于存在诸如数据缺失等技术假象,这些表达状态通常噪声更大。虽然存在许多用于推断基因调控网络的算法,但其中很少有算法能够利用单细胞表达数据中存在的额外表达状态,同时又不受大量技术噪声的不利影响。
在此,我们介绍了BTR,这是一种使用新颖的布尔状态空间评分函数,利用单细胞表达数据训练异步布尔模型的算法。BTR能够通过改善模型预测与表达数据之间的匹配度来优化现有的布尔模型并重建新的布尔模型。我们证明,对于贝叶斯网络,布尔评分函数的表现优于贝叶斯信息准则(BIC)评分函数。此外,我们表明,在批量和单细胞合成表达数据中,BTR均优于许多其他网络推断算法。最后,我们介绍了两个案例研究,其中我们使用BTR来改进已发表的布尔模型,以便产生潜在的新生物学见解。
BTR提供了一种使用单细胞表达数据优化或重建布尔模型的新方法。布尔模型对于使用单细胞数据进行网络重建特别有用,因为它对数据缺失的影响更具鲁棒性。此外,BTR不假设细胞间表达状态存在任何关系,它有助于在尽可能少的假设下重建基因调控网络。鉴于布尔模型的简单性以及生物学家对单细胞基因组学的迅速采用,BTR有可能在生物医学研究的许多领域产生影响。