Zhou Xiaoyuan, Nardini Christine
Group of Clinical Genomic Networks, Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Shanghai, People's Republic of China.
University of Chinese Academy of Sciences, Beijing, People's Republic of China.
BMC Syst Biol. 2016 Nov 15;10(1):107. doi: 10.1186/s12918-016-0344-6.
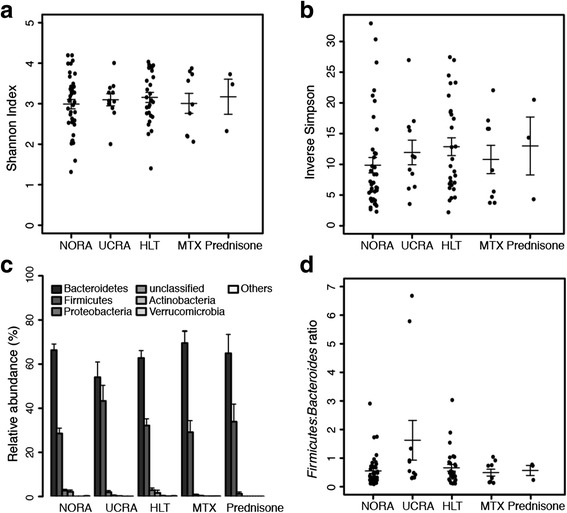
Sequencing technologies applied to mammals' microbiomes have revolutionized our understanding of health and disease. Hence, to assess diseases' progression as well as therapies longterm effects, the impact of maladies and drugs on the gut-intestinal (GI) microbiome has to be evaluated. Typical metagenomic analyses are run to associate to a condition (disease, therapy, diet) a pool of bacteria, whose eubiotic/dysbiotic potential is assessed either by α-diversity, a measure of the varieties populating the microbiome, or by Firmicutes to Bacteroides ratio, associated to systemic inflammation, and finally by manual and direct inspection of bacteria's biological functions, when known. These approaches lead to results sometimes difficult to interpret in terms of the evolution towards a specific microbial composition, harmed by large areas of unknown.
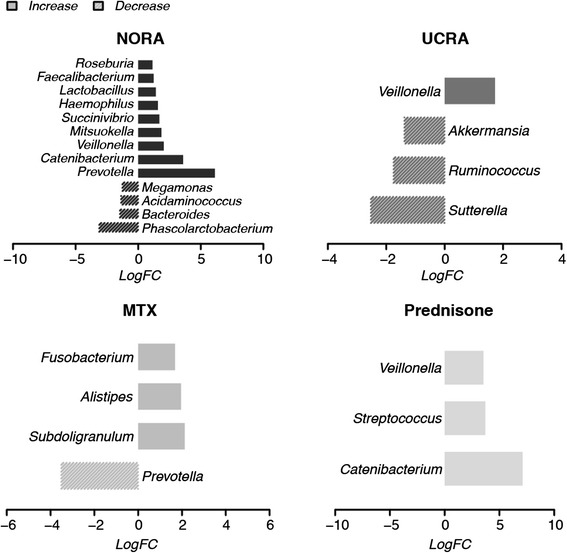
We propose to additionally evaluate a microbiome based on its global composition, by automatic annotation of pathogenic genera and statistical assessment of the net varied frequency of harmless versus harmful organisms. This application is intuitive, quantitative and computationally efficient and designed to cope with the currently incomplete species' functional knowledge. Our results, applied to human GI-microbiome data exemplify how this layer of information provides additional insights into treatments' impact on the GI microbiome, allowing to characterize a more physiologic effects of Prednisone versus Methotrexate, two treatments for rheumatoid arthritis (RA) a complex autoimmune systemic disease.
Our quantitative analysis integrates with previous approaches offering an additional systemic level of interpretation here applied, for its potential to translate into clinically relevant information, to the therapies for RA.
应用于哺乳动物微生物群的测序技术彻底改变了我们对健康和疾病的理解。因此,为了评估疾病的进展以及治疗的长期效果,必须评估疾病和药物对胃肠道(GI)微生物群的影响。典型的宏基因组分析是将一组细菌与一种状况(疾病、治疗、饮食)相关联,这些细菌的有益/失调潜力通过α多样性(一种衡量微生物群中种类的指标)、与全身炎症相关的厚壁菌门与拟杆菌门的比例,以及在已知细菌生物学功能时通过人工和直接检查来评估。这些方法有时难以从向特定微生物组成的演变角度进行解释,因为存在大片未知区域。
我们建议通过自动注释致病属以及对无害与有害生物体的净变化频率进行统计评估,来额外评估基于其整体组成的微生物群。该应用直观、定量且计算效率高,旨在应对目前物种功能知识不完整的情况。我们将结果应用于人类胃肠道微生物群数据,举例说明了这一层信息如何为治疗对胃肠道微生物群的影响提供额外的见解,从而能够表征泼尼松与甲氨蝶呤这两种类风湿关节炎(RA,一种复杂的自身免疫性全身性疾病)治疗方法更生理的效果。
我们的定量分析与先前的方法相结合,在此提供了一个额外的系统解释层面,因其有可能转化为临床相关信息,应用于类风湿关节炎的治疗。