Gluck Christian, Min Sangwon, Oyelakin Akinsola, Smalley Kirsten, Sinha Satrajit, Romano Rose-Anne
Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY, 14203, USA.
Department of Oral Biology, School of Dental Medicine, State University of New York at Buffalo, 3435 Main Street, Buffalo, NY, 14214, USA.
BMC Genomics. 2016 Nov 16;17(1):923. doi: 10.1186/s12864-016-3228-7.
Mouse models have served a valuable role in deciphering various facets of Salivary Gland (SG) biology, from normal developmental programs to diseased states. To facilitate such studies, gene expression profiling maps have been generated for various stages of SG organogenesis. However these prior studies fall short of capturing the transcriptional complexity due to the limited scope of gene-centric microarray-based technology. Compared to microarray, RNA-sequencing (RNA-seq) offers unbiased detection of novel transcripts, broader dynamic range and high specificity and sensitivity for detection of genes, transcripts, and differential gene expression. Although RNA-seq data, particularly under the auspices of the ENCODE project, have covered a large number of biological specimens, studies on the SG have been lacking.
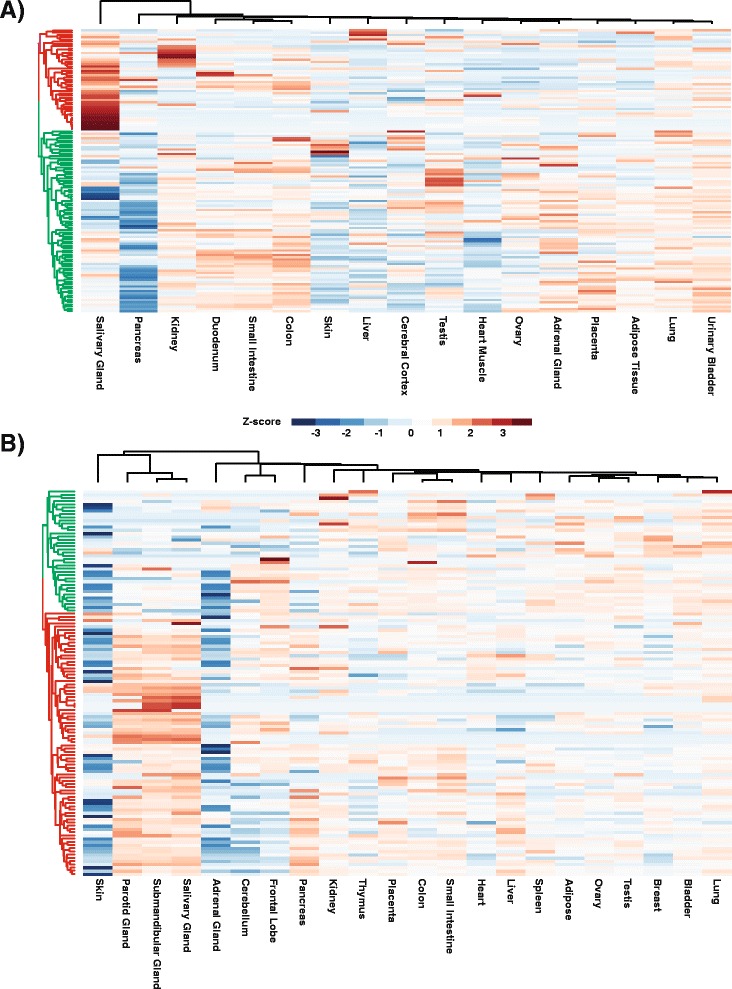
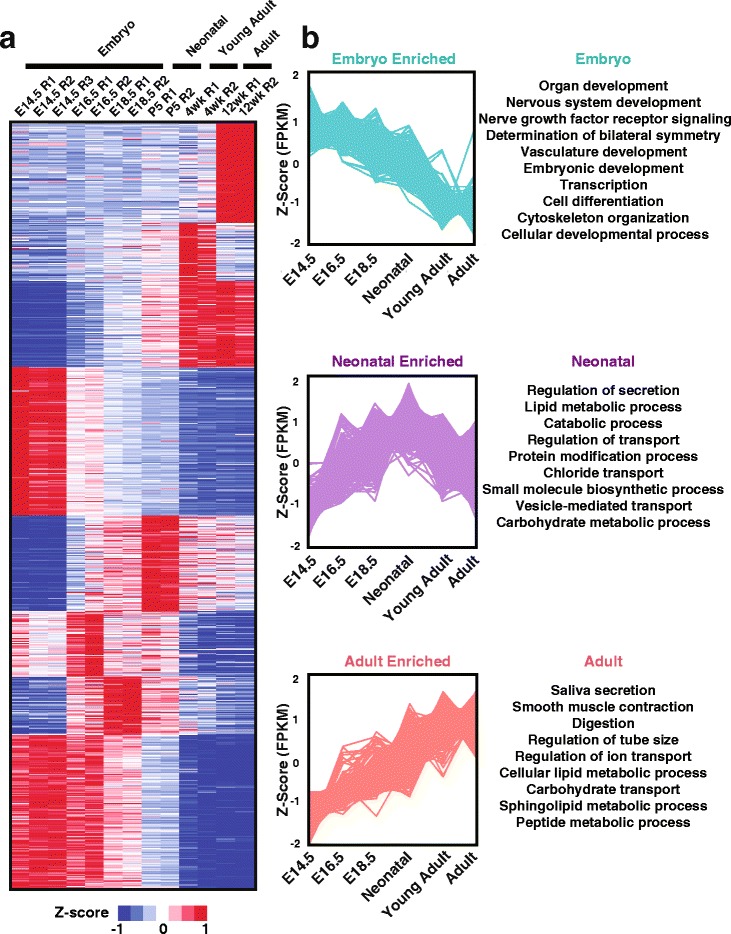
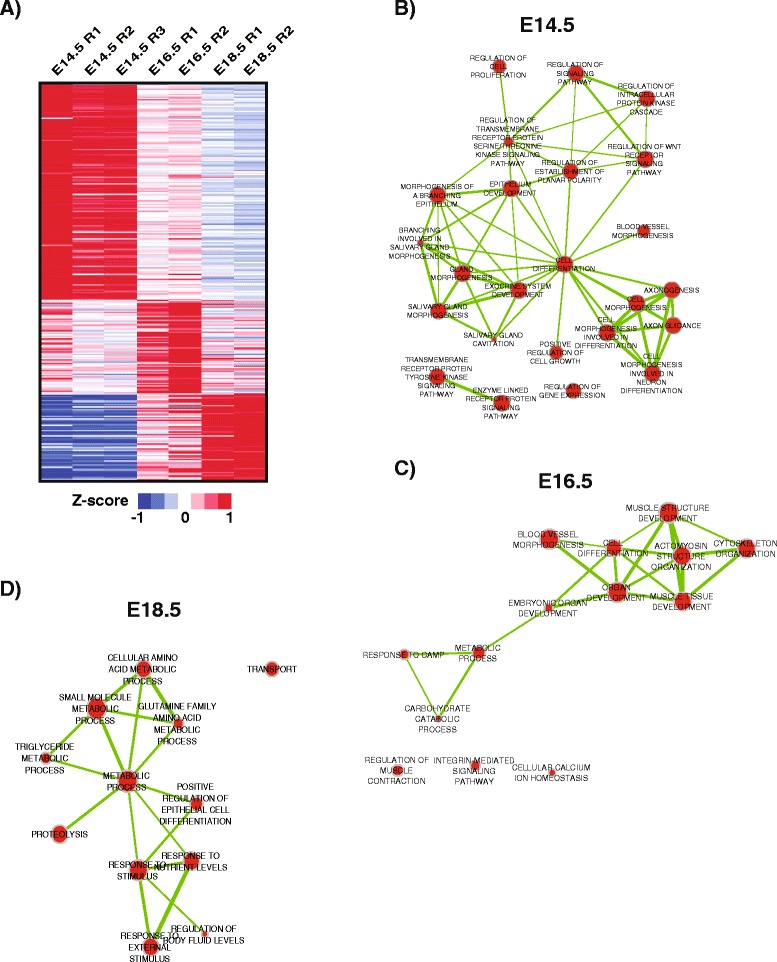
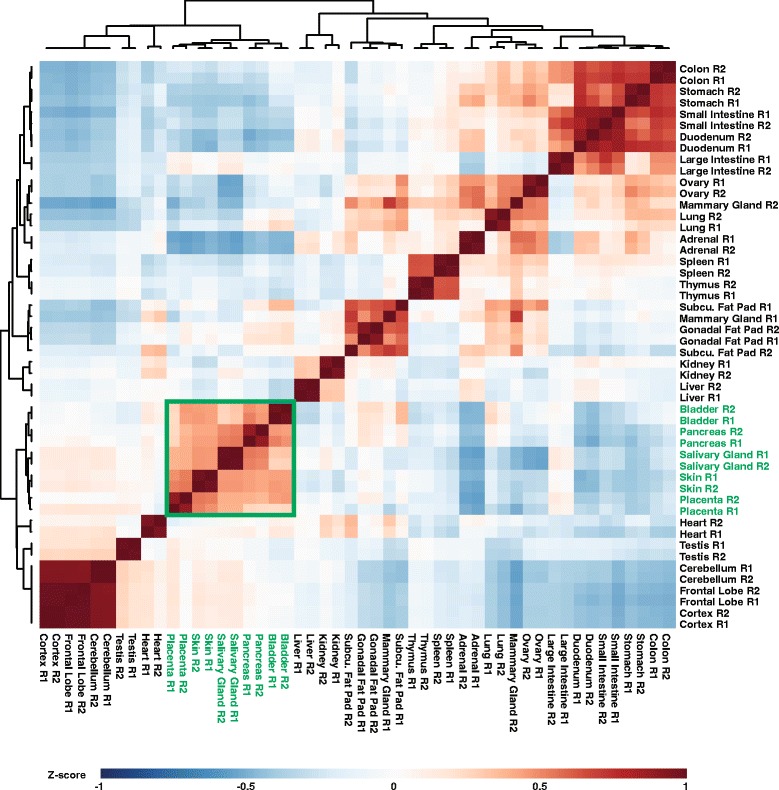
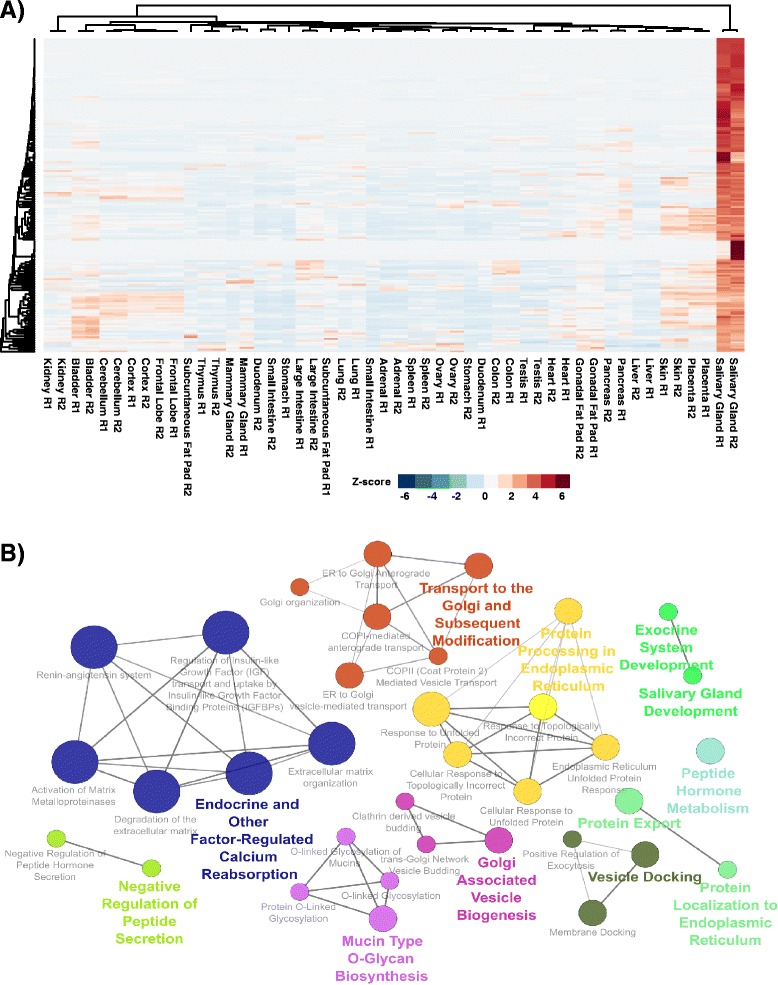
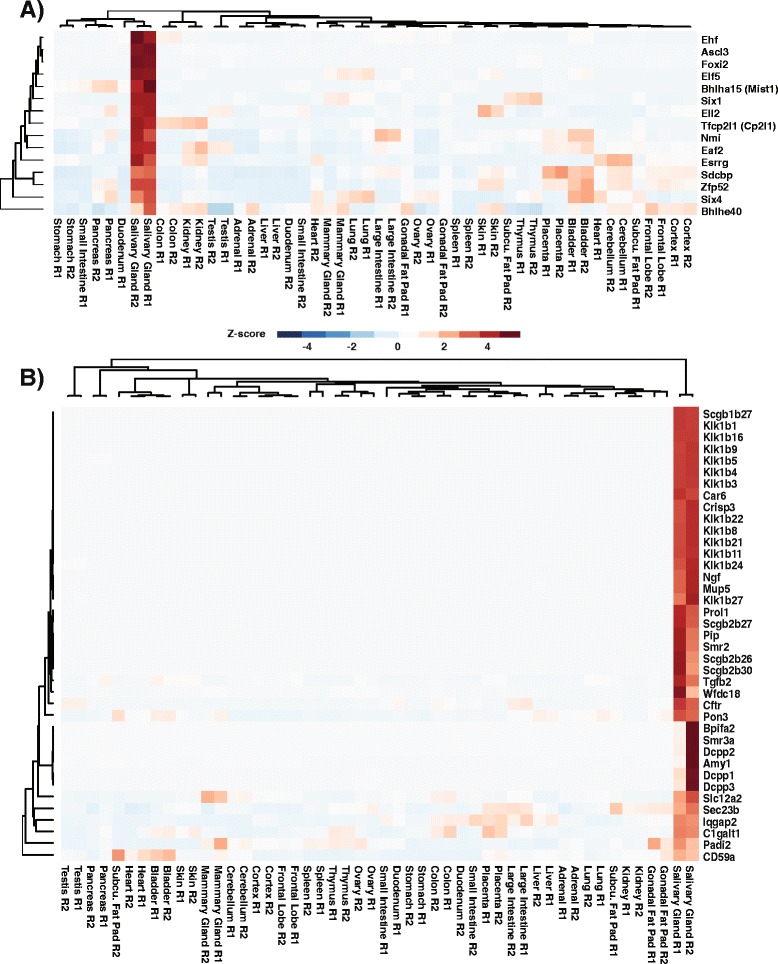
To better appreciate the wide spectrum of gene expression profiles, we isolated RNA from mouse submandibular salivary glands at different embryonic and adult stages. In parallel, we processed RNA-seq data for 24 organs and tissues obtained from the mouse ENCODE consortium and calculated the average gene expression values. To identify molecular players and pathways likely to be relevant for SG biology, we performed functional gene enrichment analysis, network construction and hierarchal clustering of the RNA-seq datasets obtained from different stages of SG development and maturation, and other mouse organs and tissues. Our bioinformatics-based data analysis not only reaffirmed known modulators of SG morphogenesis but revealed novel transcription factors and signaling pathways unique to mouse SG biology and function. Finally we demonstrated that the unique SG gene signature obtained from our mouse studies is also well conserved and can demarcate features of the human SG transcriptome that is different from other tissues.
Our RNA-seq based Atlas has revealed a high-resolution cartographic view of the dynamic transcriptomic landscape of the mouse SG at various stages. These RNA-seq datasets will complement pre-existing microarray based datasets, including the Salivary Gland Molecular Anatomy Project by offering a broader systems-biology based perspective rather than the classical gene-centric view. Ultimately such resources will be valuable in providing a useful toolkit to better understand how the diverse cell population of the SG are organized and controlled during development and differentiation.
小鼠模型在解析唾液腺(SG)生物学的各个方面发挥了重要作用,从正常发育程序到疾病状态。为便于此类研究,已针对SG器官发生的各个阶段生成了基因表达谱图。然而,由于基于基因芯片技术的局限性,这些先前的研究未能捕捉到转录复杂性。与芯片相比,RNA测序(RNA-seq)能够无偏地检测新转录本,具有更宽的动态范围以及对基因、转录本和差异基因表达检测的高特异性和敏感性。尽管RNA-seq数据,特别是在ENCODE项目的支持下,已涵盖了大量生物样本,但关于SG的研究仍然匮乏。
为了更好地了解广泛的基因表达谱,我们从小鼠不同胚胎期和成年期的下颌下唾液腺中分离RNA。同时,我们处理了从小鼠ENCODE联盟获得的24种器官和组织的RNA-seq数据,并计算了平均基因表达值。为了识别可能与SG生物学相关的分子参与者和途径,我们对从SG发育和成熟的不同阶段以及其他小鼠器官和组织获得的RNA-seq数据集进行了功能基因富集分析、网络构建和层次聚类。我们基于生物信息学的数据分析不仅再次确认了已知的SG形态发生调节因子,还揭示了小鼠SG生物学和功能特有的新转录因子和信号通路。最后,我们证明从我们的小鼠研究中获得的独特SG基因特征也具有良好的保守性,并且可以区分人类SG转录组与其他组织不同的特征。
我们基于RNA-seq的图谱揭示了小鼠SG在各个阶段动态转录组景观的高分辨率制图视图。这些RNA-seq数据集将补充现有的基于芯片的数据集,包括唾液腺分子解剖项目,通过提供更广泛的基于系统生物学的视角,而不是传统的以基因为中心的观点。最终,这些资源将有助于提供一个有用的工具包,以更好地了解SG中不同细胞群体在发育和分化过程中是如何组织和调控的。