Liu Li, Chang Yung, Yang Tao, Noren David P, Long Byron, Kornblau Steven, Qutub Amina, Ye Jieping
Department of Biomedical Informatics Arizona State University Tempe AZ USA.
School of Life Science Arizona State University Tempe AZ USA.
Evol Appl. 2016 Oct 21;10(1):68-76. doi: 10.1111/eva.12417. eCollection 2017 Jan.
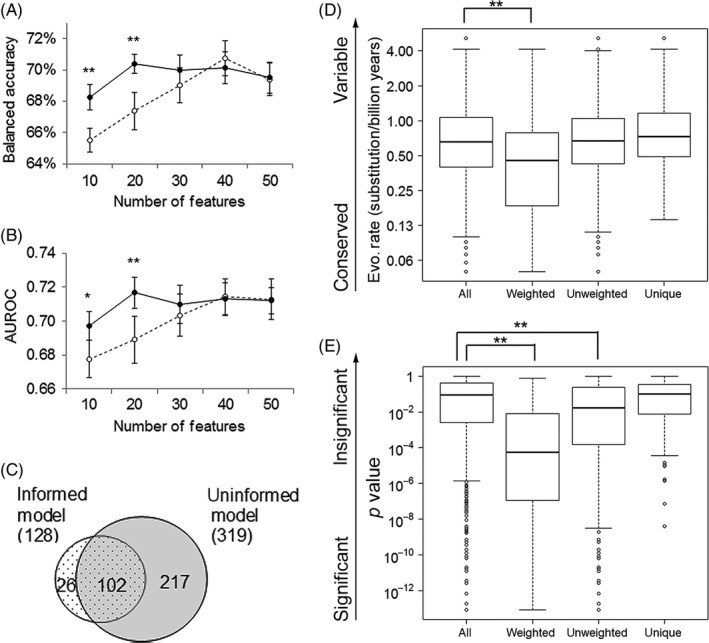
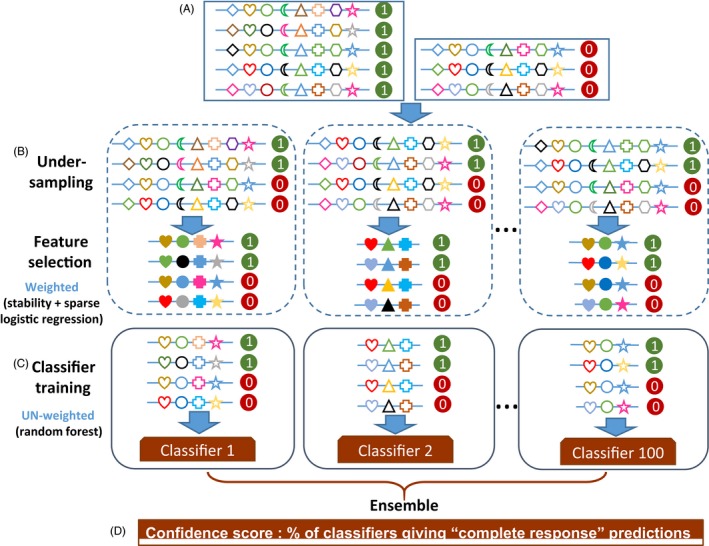
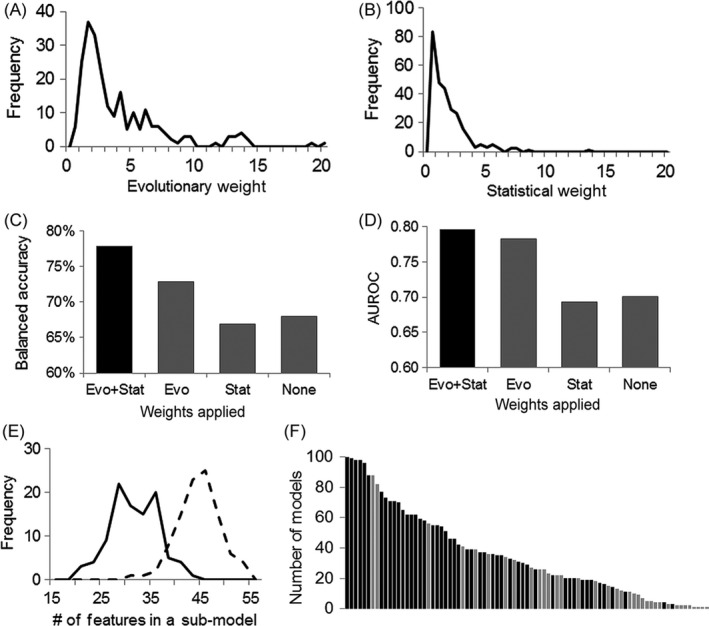
Despite wide applications of high-throughput biotechnologies in cancer research, many biomarkers discovered by exploring large-scale omics data do not provide satisfactory performance when used to predict cancer treatment outcomes. This problem is partly due to the overlooking of functional implications of molecular markers. Here, we present a novel computational method that uses evolutionary conservation as prior knowledge to discover bona fide biomarkers. Evolutionary selection at the molecular level is nature's test on functional consequences of genetic elements. By prioritizing genes that show significant statistical association and high functional impact, our new method reduces the chances of including spurious markers in the predictive model. When applied to predicting therapeutic responses for patients with acute myeloid leukemia and to predicting metastasis for patients with prostate cancers, the new method gave rise to evolution-informed models that enjoyed low complexity and high accuracy. The identified genetic markers also have significant implications in tumor progression and embrace potential drug targets. Because evolutionary conservation can be estimated as a gene-specific, position-specific, or allele-specific parameter on the nucleotide level and on the protein level, this new method can be extended to apply to miscellaneous "omics" data to accelerate biomarker discoveries.
尽管高通量生物技术在癌症研究中得到了广泛应用,但通过探索大规模组学数据发现的许多生物标志物在用于预测癌症治疗结果时,其表现并不令人满意。这个问题部分归因于对分子标记功能意义的忽视。在此,我们提出了一种新颖的计算方法,该方法利用进化保守性作为先验知识来发现真正的生物标志物。分子水平上的进化选择是自然对遗传元件功能后果的检验。通过对显示出显著统计关联和高功能影响的基因进行优先级排序,我们的新方法降低了在预测模型中纳入虚假标记的可能性。当应用于预测急性髓系白血病患者的治疗反应以及预测前列腺癌患者的转移情况时,新方法产生了具有低复杂度和高精度的基于进化信息的模型。所鉴定的遗传标记在肿瘤进展中也具有重要意义,并包含潜在的药物靶点。由于进化保守性可以在核苷酸水平和蛋白质水平上作为基因特异性、位置特异性或等位基因特异性参数进行估计,因此这种新方法可以扩展应用于各种“组学”数据,以加速生物标志物的发现。