Institute for Molecular Medicine Finland (FIMM), University of Helsinki, 00290 Helsinki, Finland.
Helsinki Institute for Information Technology (HIIT), Aalto University, 02150 Espoo, Finland.
Bioinformatics. 2018 Apr 15;34(8):1353-1362. doi: 10.1093/bioinformatics/btx766.
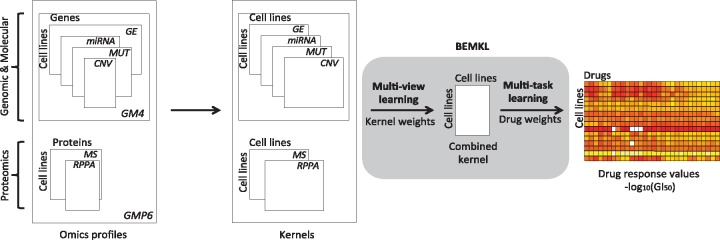
Proteomics profiling is increasingly being used for molecular stratification of cancer patients and cell-line panels. However, systematic assessment of the predictive power of large-scale proteomic technologies across various drug classes and cancer types is currently lacking. To that end, we carried out the first pan-cancer, multi-omics comparative analysis of the relative performance of two proteomic technologies, targeted reverse phase protein array (RPPA) and global mass spectrometry (MS), in terms of their accuracy for predicting the sensitivity of cancer cells to both cytotoxic chemotherapeutics and molecularly targeted anticancer compounds.
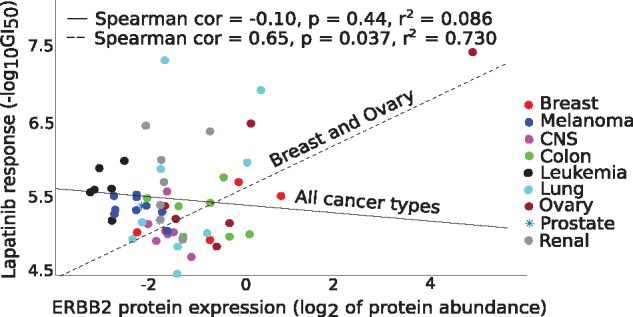
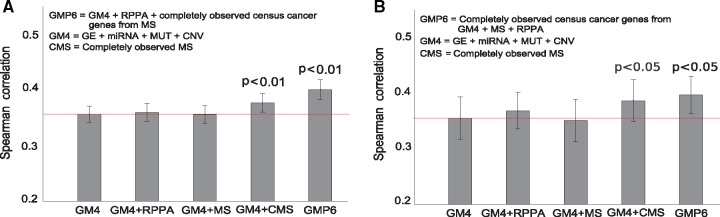
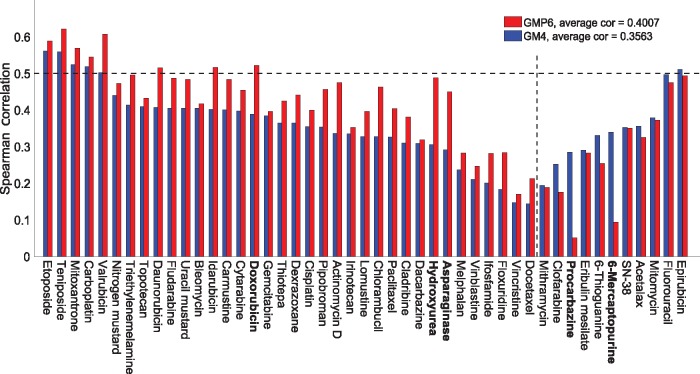
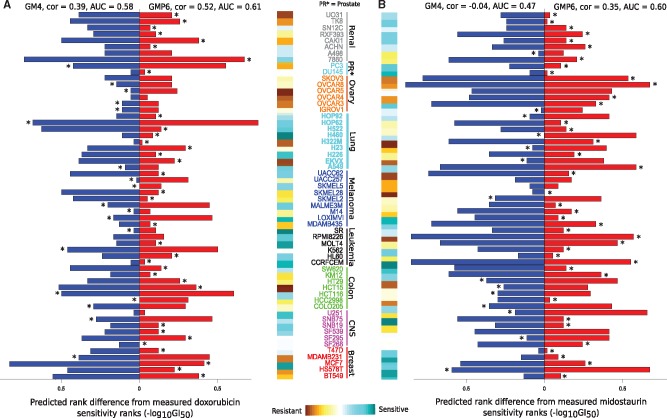
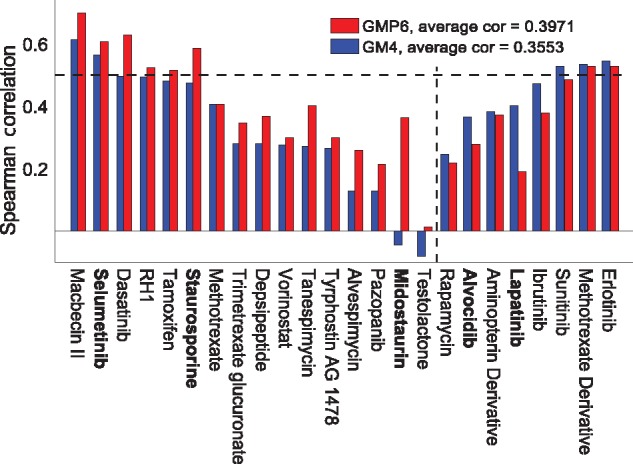
Our results in two cell-line panels demonstrate how MS profiling improves drug response predictions beyond that of the RPPA or the other omics profiles when used alone. However, frequent missing MS data values complicate its use in predictive modeling and required additional filtering, such as focusing on completely measured or known oncoproteins, to obtain maximal predictive performance. Rather strikingly, the two proteomics profiles provided complementary predictive signal both for the cytotoxic and targeted compounds. Further, information about the cellular-abundance of primary target proteins was found critical for predicting the response of targeted compounds, although the non-target features also contributed significantly to the predictive power. The clinical relevance of the selected protein markers was confirmed in cancer patient data. These results provide novel insights into the relative performance and optimal use of the widely applied proteomic technologies, MS and RPPA, which should prove useful in translational applications, such as defining the best combination of omics technologies and marker panels for understanding and predicting drug sensitivities in cancer patients.
Processed datasets, R as well as Matlab implementations of the methods are available at https://github.com/mehr-een/bemkl-rbps.
mehreen.ali@helsinki.fi or tero.aittokallio@fimm.fi.
Supplementary data are available at Bioinformatics online.
蛋白质组学分析越来越多地被用于癌症患者和细胞系的分子分层。然而,目前缺乏对各种药物类别和癌症类型的大规模蛋白质组学技术的预测能力进行系统评估。为此,我们进行了第一次泛癌症、多组学比较分析,比较了两种蛋白质组学技术(靶向反相蛋白阵列(RPPA)和全局质谱(MS))在预测癌细胞对细胞毒性化疗药物和分子靶向抗癌化合物敏感性方面的相对性能。
我们在两个细胞系面板中的结果表明,当单独使用时,MS 分析如何通过其对药物反应的预测能力来改善 RPPA 或其他组学分析的预测。然而,频繁出现的 MS 数据值缺失使得其在预测模型中使用变得复杂,需要额外的过滤,例如关注完全测量或已知的癌蛋白,以获得最大的预测性能。相当惊人的是,这两种蛋白质组学分析为细胞毒性和靶向化合物都提供了互补的预测信号。此外,关于主要靶蛋白在细胞内丰度的信息被发现对预测靶向化合物的反应至关重要,尽管非靶特征也对预测能力有很大贡献。在癌症患者数据中验证了所选蛋白质标记物的临床相关性。这些结果为广泛应用的蛋白质组学技术(MS 和 RPPA)的相对性能和最佳使用提供了新的见解,这将有助于转化应用,例如确定最佳的组学技术和标记物组合,以了解和预测癌症患者的药物敏感性。
处理后的数据集、R 以及 Matlab 实现方法可在 https://github.com/mehr-een/bemkl-rbps 上获得。
mehreen.ali@helsinki.fi 或 tero.aittokallio@fimm.fi。
补充数据可在生物信息学在线获得。