Odhams Christopher A, Cortini Andrea, Chen Lingyan, Roberts Amy L, Viñuela Ana, Buil Alfonso, Small Kerrin S, Dermitzakis Emmanouil T, Morris David L, Vyse Timothy J, Cunninghame Graham Deborah S
Department of Medical & Molecular Genetics, King's College London, London, UK.
Department of Twin Research, King's College London, London, UK.
Hum Mol Genet. 2017 Mar 1;26(5):1003-1017. doi: 10.1093/hmg/ddw417.
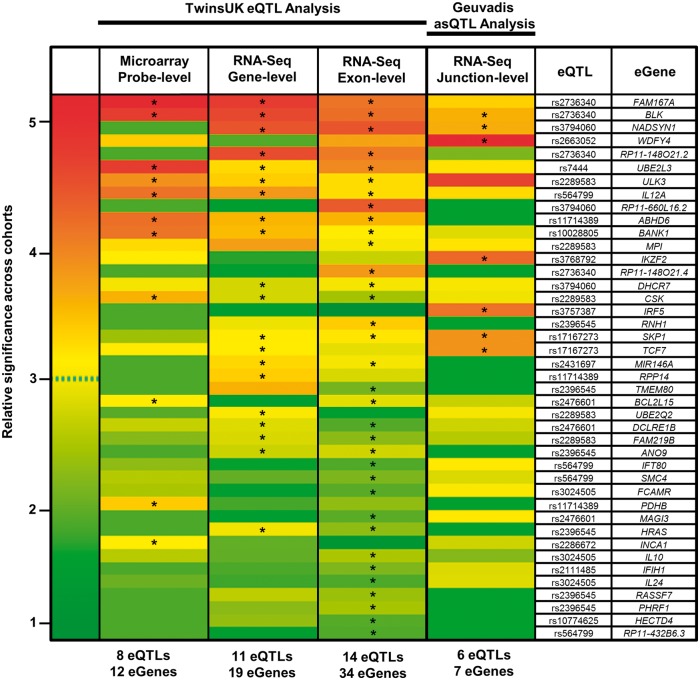
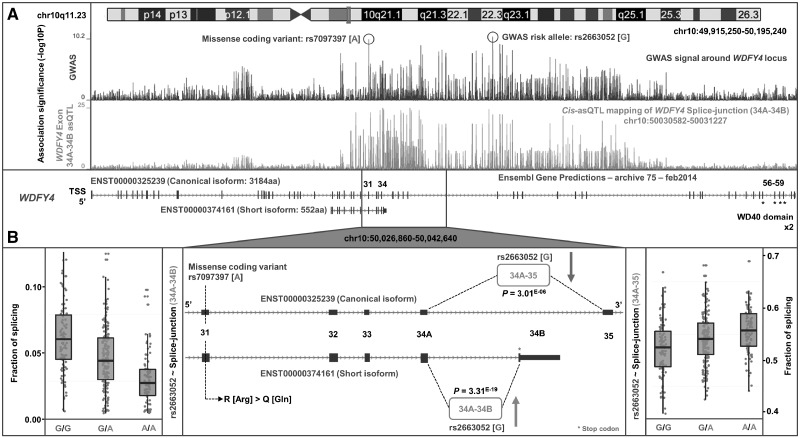
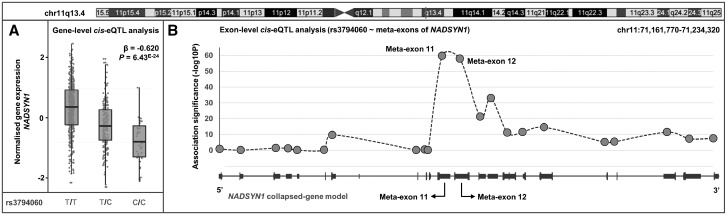
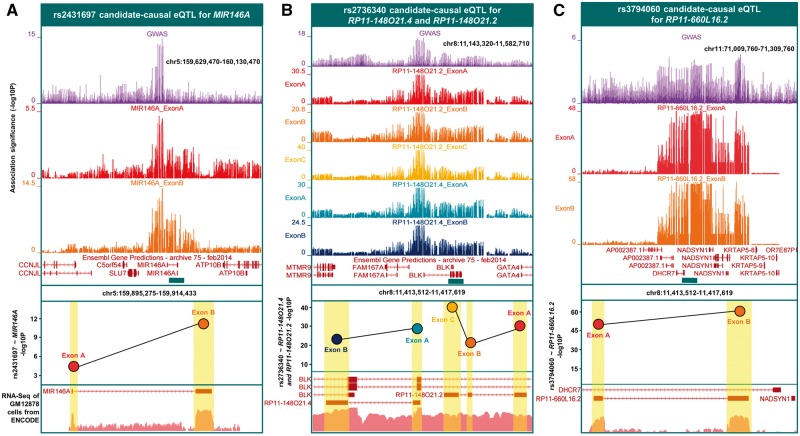
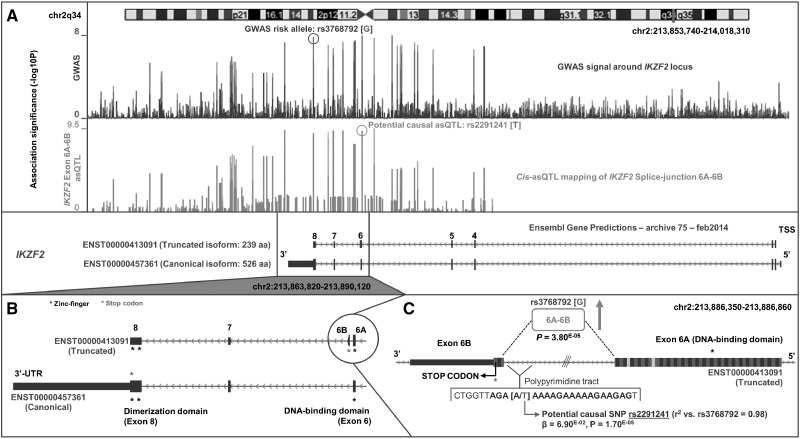
Studies attempting to functionally interpret complex-disease susceptibility loci by GWAS and eQTL integration have predominantly employed microarrays to quantify gene-expression. RNA-Seq has the potential to discover a more comprehensive set of eQTLs and illuminate the underlying molecular consequence. We examine the functional outcome of 39 variants associated with Systemic Lupus Erythematosus (SLE) through the integration of GWAS and eQTL data from the TwinsUK microarray and RNA-Seq cohort in lymphoblastoid cell lines. We use conditional analysis and a Bayesian colocalisation method to provide evidence of a shared causal-variant, then compare the ability of each quantification type to detect disease relevant eQTLs and eGenes. We discovered the greatest frequency of candidate-causal eQTLs using exon-level RNA-Seq, and identified novel SLE susceptibility genes (e.g. NADSYN1 and TCF7) that were concealed using microarrays, including four non-coding RNAs. Many of these eQTLs were found to influence the expression of several genes, supporting the notion that risk haplotypes may harbour multiple functional effects. Novel SLE associated splicing events were identified in the T-reg restricted transcription factor, IKZF2, and other candidate genes (e.g. WDFY4) through asQTL mapping using the Geuvadis cohort. We have significantly increased our understanding of the genetic control of gene-expression in SLE by maximising the leverage of RNA-Seq and performing integrative GWAS-eQTL analysis against gene, exon, and splice-junction quantifications. We conclude that to better understand the true functional consequence of regulatory variants, quantification by RNA-Seq should be performed at the exon-level as a minimum, and run in parallel with gene and splice-junction level quantification.
试图通过全基因组关联研究(GWAS)和表达数量性状基因座(eQTL)整合对复杂疾病易感位点进行功能解释的研究,主要采用微阵列来定量基因表达。RNA测序(RNA-Seq)有潜力发现更全面的eQTL集,并阐明潜在的分子后果。我们通过整合来自TwinsUK微阵列和淋巴母细胞系RNA-Seq队列的GWAS和eQTL数据,研究了39个与系统性红斑狼疮(SLE)相关的变异的功能结果。我们使用条件分析和贝叶斯共定位方法来提供共享因果变异的证据,然后比较每种定量类型检测与疾病相关的eQTL和e基因的能力。我们使用外显子水平的RNA-Seq发现了候选因果eQTL的最高频率,并鉴定出了使用微阵列时被隐藏的新型SLE易感基因(如NADSYN1和TCF7),包括四个非编码RNA。发现许多这些eQTL会影响多个基因的表达,支持了风险单倍型可能具有多种功能效应的观点。通过使用Geuvadis队列进行剪接数量性状基因座(asQTL)定位,在T细胞调节限制转录因子IKZF2和其他候选基因(如WDFY4)中鉴定出了新型SLE相关剪接事件。通过最大限度地利用RNA-Seq并针对基因、外显子和剪接连接定量进行综合GWAS-eQTL分析,我们显著增进了对SLE中基因表达遗传控制的理解。我们得出结论,为了更好地理解调控变异的真正功能后果,至少应在外显子水平进行RNA-Seq定量,并与基因和剪接连接水平定量并行进行。