Single-Cell Center, Shandong Key Laboratory of Energy Genetics and CAS Key Laboratory of Biofuels, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences. Qingdao, Shandong, 266101, China.
Sci Rep. 2017 Jan 12;7:40371. doi: 10.1038/srep40371.
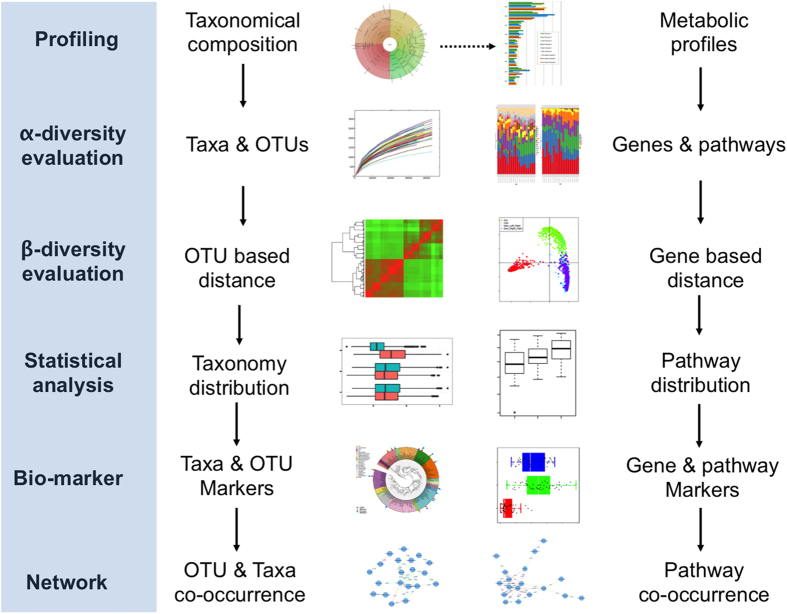
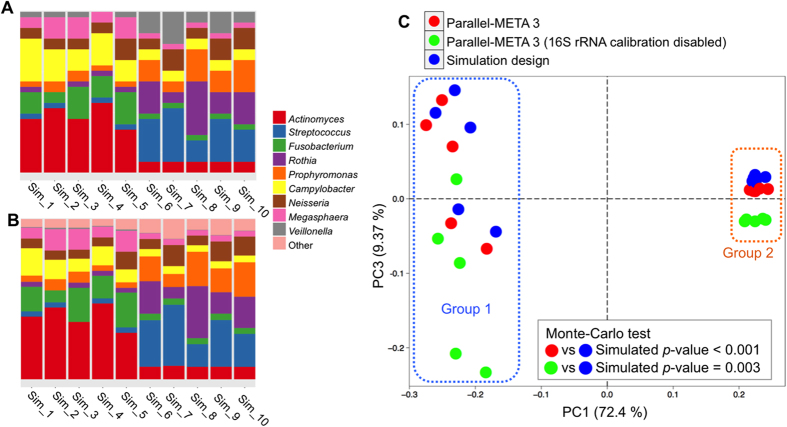
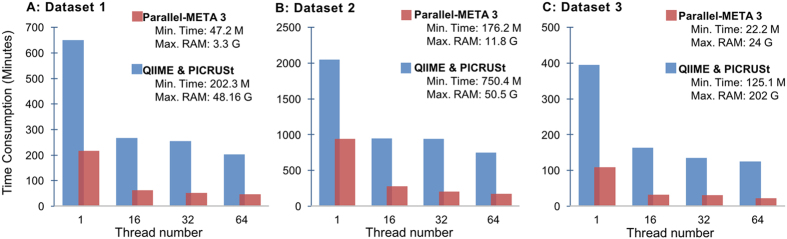
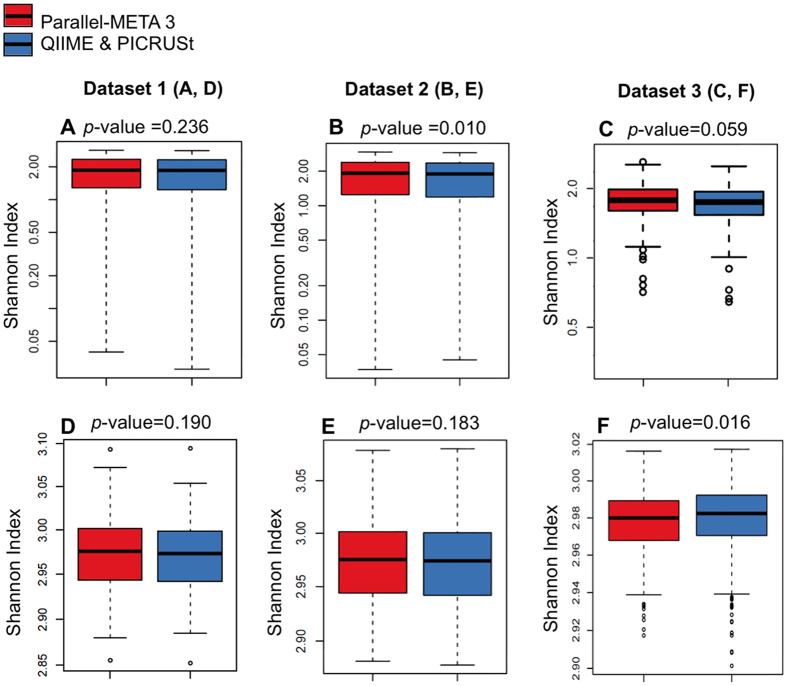
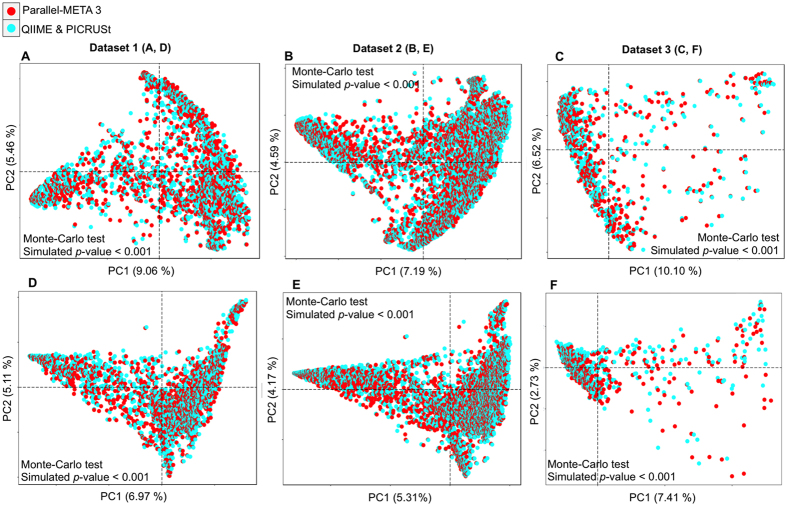
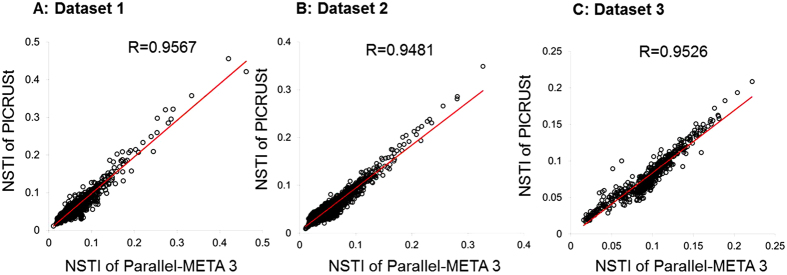
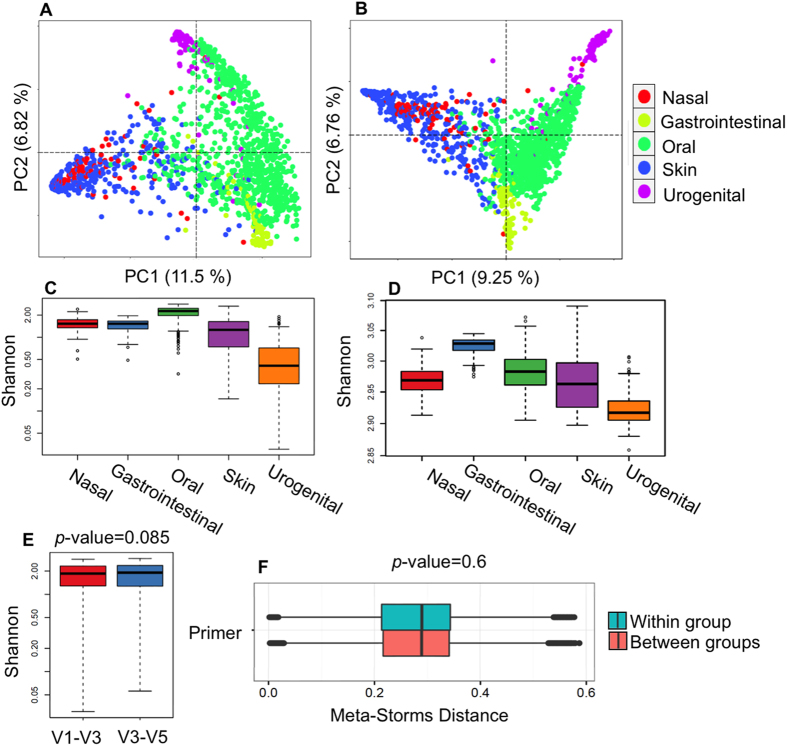
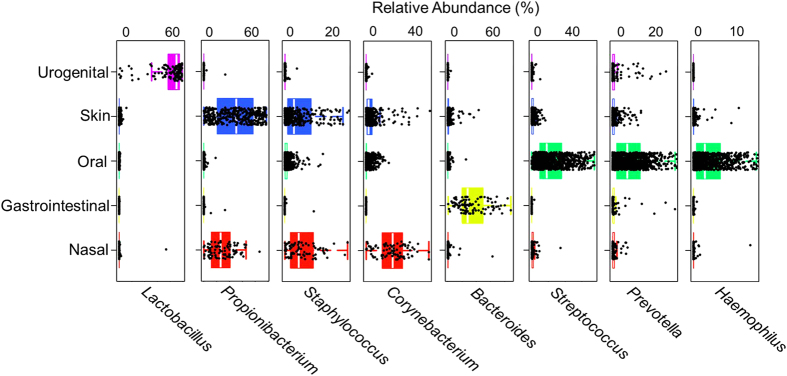
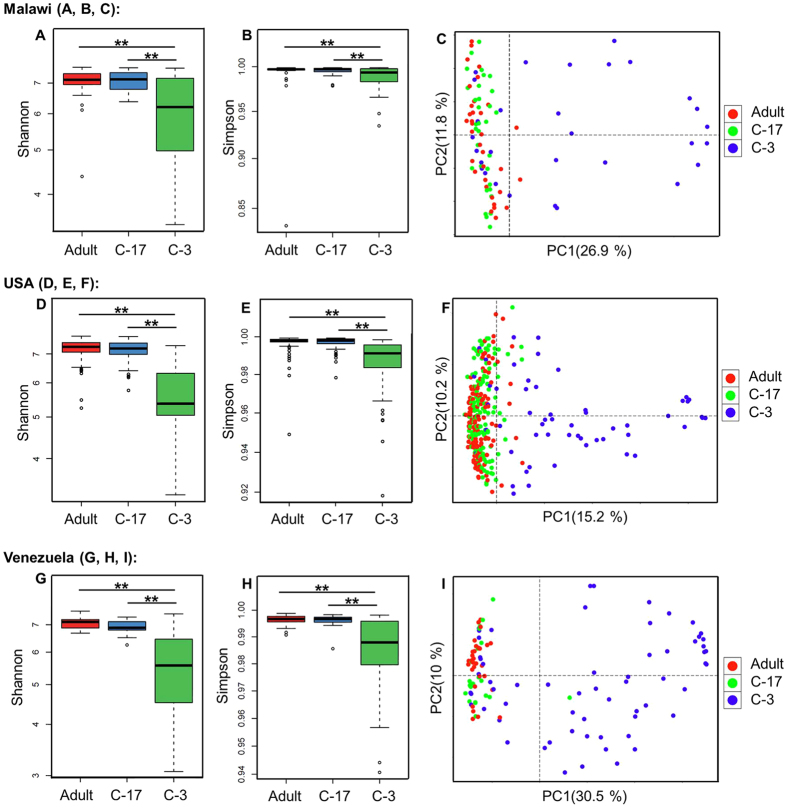
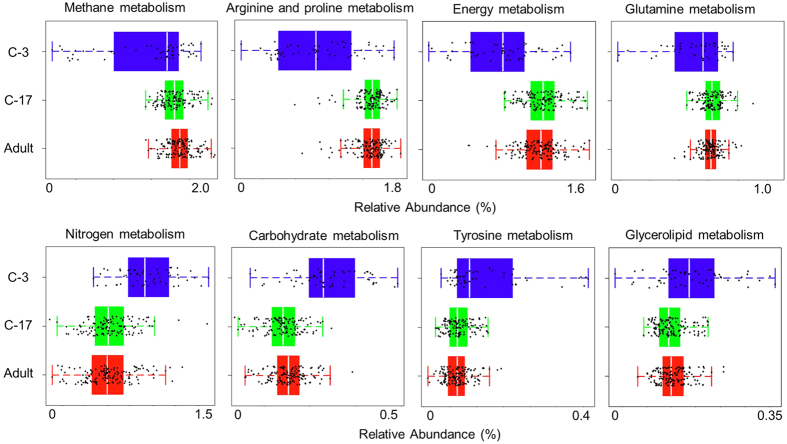
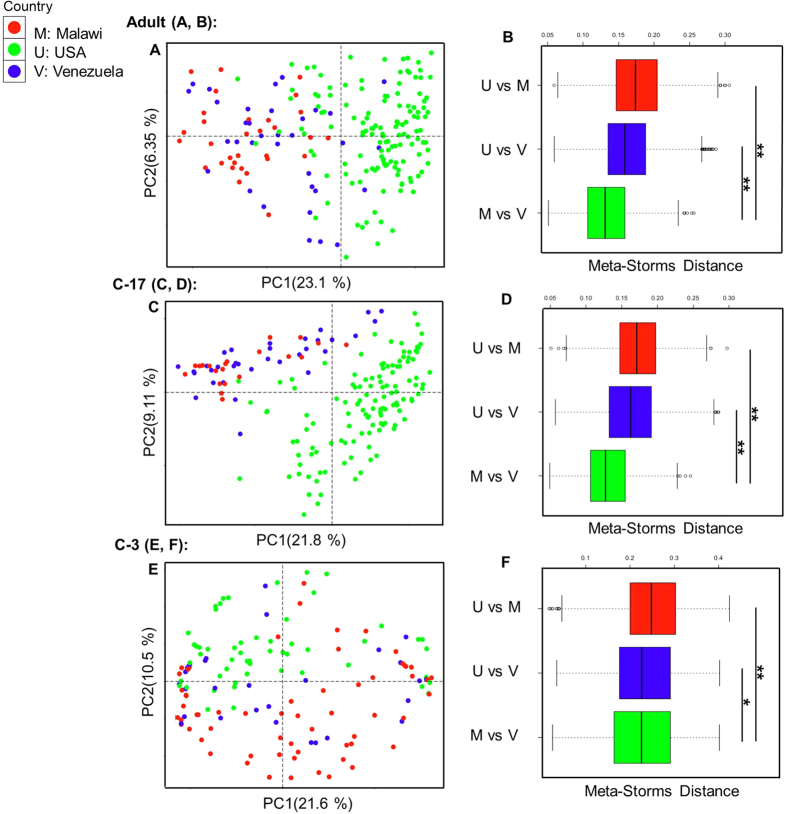
The number of metagenomes is increasing rapidly. However, current methods for metagenomic analysis are limited by their capability for in-depth data mining among a large number of microbiome each of which carries a complex community structure. Moreover, the complexity of configuring and operating computational pipeline also hinders efficient data processing for the end users. In this work we introduce Parallel-META 3, a comprehensive and fully automatic computational toolkit for rapid data mining among metagenomic datasets, with advanced features including 16S rRNA extraction for shotgun sequences, 16S rRNA copy number calibration, 16S rRNA based functional prediction, diversity statistics, bio-marker selection, interaction network construction, vector-graph-based visualization and parallel computing. Application of Parallel-META 3 on 5,337 samples with 1,117,555,208 sequences from diverse studies and platforms showed it could produce similar results as QIIME and PICRUSt with much faster speed and lower memory usage, which demonstrates its ability to unravel the taxonomical and functional dynamics patterns across large datasets and elucidate ecological links between microbiome and the environment. Parallel-META 3 is implemented in C/C++ and R, and integrated into an executive package for rapid installation and easy access under Linux and Mac OS X. Both binary and source code packages are available at http://bioinfo.single-cell.cn/parallel-meta.html.
宏基因组的数量正在迅速增加。然而,目前的宏基因组分析方法受到其在大量微生物组中进行深度数据挖掘的能力的限制,而每个微生物组都具有复杂的群落结构。此外,配置和操作计算管道的复杂性也阻碍了最终用户对高效数据处理。在这项工作中,我们引入了 Parallel-META 3,这是一个用于快速挖掘宏基因组数据集的综合和全自动计算工具包,具有先进的功能,包括用于 shotgun 序列的 16S rRNA 提取、16S rRNA 拷贝数校准、基于 16S rRNA 的功能预测、多样性统计、生物标志物选择、相互作用网络构建、基于向量图的可视化和并行计算。在来自不同研究和平台的 5337 个样本和 1117555208 个序列上应用 Parallel-META 3 的结果表明,它可以与 QIIME 和 PICRUSt 产生类似的结果,但速度更快,内存使用量更低,这证明了它能够揭示大型数据集的分类和功能动态模式,并阐明微生物组与环境之间的生态联系。Parallel-META 3 是用 C/C++和 R 实现的,并集成到一个执行包中,以便在 Linux 和 Mac OS X 下快速安装和轻松访问。二进制和源代码包均可在 http://bioinfo.single-cell.cn/parallel-meta.html 获得。