Lai Yi-Pin, Wang Liang-Bo, Wang Wei-An, Lai Liang-Chuan, Tsai Mong-Hsun, Lu Tzu-Pin, Chuang Eric Y
Bioinformatics and Biostatistics Core, Center of Genomic Medicine, National Taiwan University, Taipei, Taiwan.
Graduate Institute of Biomedical Electronics and Bioinformatics, Department of Electrical Engineering, National Taiwan University, Taipei, Taiwan.
BMC Bioinformatics. 2017 Jan 14;18(1):35. doi: 10.1186/s12859-016-1438-2.
With the advancement in high-throughput technologies, researchers can simultaneously investigate gene expression and copy number alteration (CNA) data from individual patients at a lower cost. Traditional analysis methods analyze each type of data individually and integrate their results using Venn diagrams. Challenges arise, however, when the results are irreproducible and inconsistent across multiple platforms. To address these issues, one possible approach is to concurrently analyze both gene expression profiling and CNAs in the same individual.
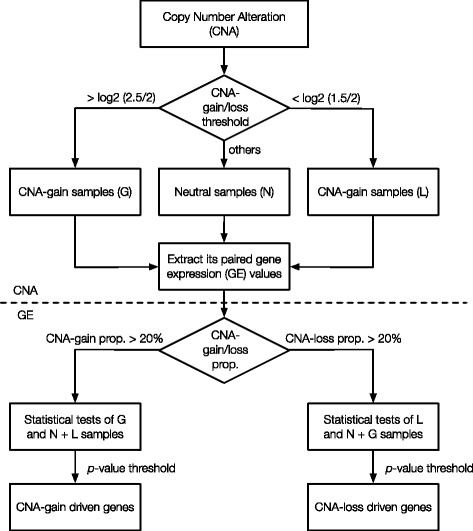
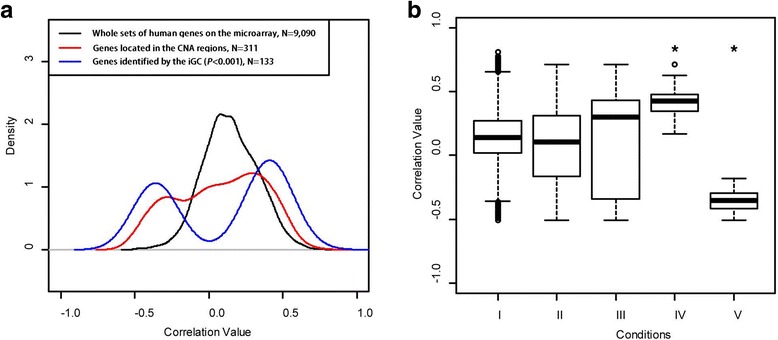
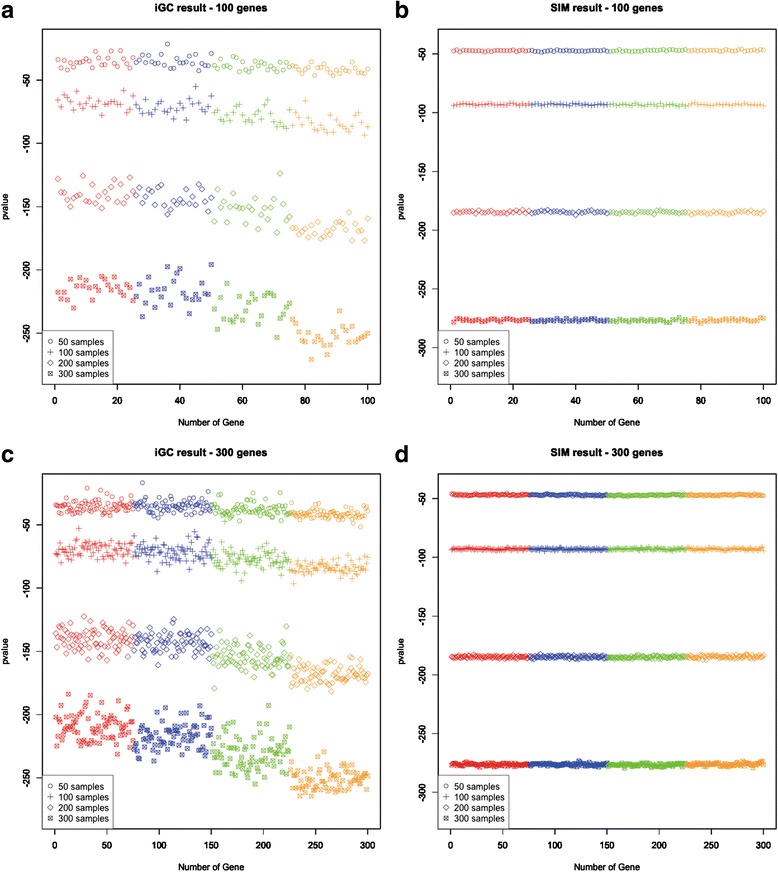
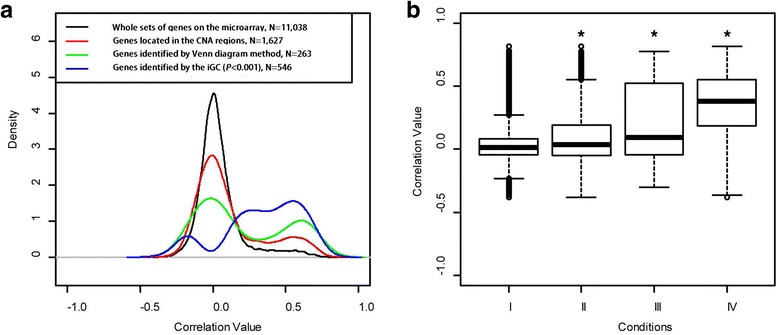
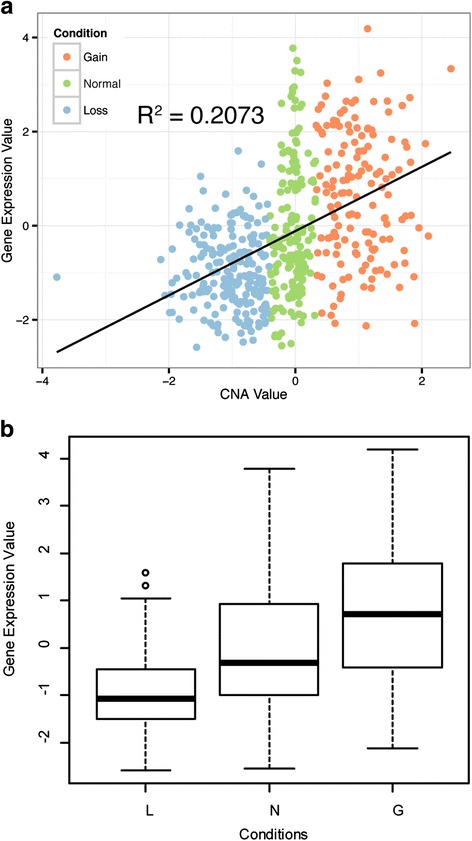
We have developed an open-source R/Bioconductor package (iGC). Multiple input formats are supported and users can define their own criteria for identifying differentially expressed genes driven by CNAs. The analysis of two real microarray datasets demonstrated that the CNA-driven genes identified by the iGC package showed significantly higher Pearson correlation coefficients with their gene expression levels and copy numbers than those genes located in a genomic region with CNA. Compared with the Venn diagram approach, the iGC package showed better performance.
The iGC package is effective and useful for identifying CNA-driven genes. By simultaneously considering both comparative genomic and transcriptomic data, it can provide better understanding of biological and medical questions. The iGC package's source code and manual are freely available at https://www.bioconductor.org/packages/release/bioc/html/iGC.html .
随着高通量技术的进步,研究人员能够以更低的成本同时研究个体患者的基因表达和拷贝数变异(CNA)数据。传统的分析方法分别分析每种类型的数据,并使用维恩图整合结果。然而,当结果在多个平台上不可重复且不一致时,就会出现挑战。为了解决这些问题,一种可能的方法是在同一个体中同时分析基因表达谱和CNA。
我们开发了一个开源的R/Bioconductor软件包(iGC)。它支持多种输入格式,用户可以定义自己的标准来识别由CNA驱动的差异表达基因。对两个真实微阵列数据集的分析表明,与位于存在CNA的基因组区域中的基因相比,iGC软件包识别出的由CNA驱动的基因与其基因表达水平和拷贝数显示出显著更高的皮尔逊相关系数。与维恩图方法相比,iGC软件包表现更好。
iGC软件包对于识别由CNA驱动的基因是有效且有用的。通过同时考虑比较基因组和转录组数据,它可以更好地理解生物学和医学问题。iGC软件包的源代码和手册可在https://www.bioconductor.org/packages/release/bioc/html/iGC.html免费获取。