Dalkas Georgios A, Rooman Marianne
BioModeling, BioInformatics & BioProcesses (3BIO), Université Libre de Bruxelles (ULB), CP 165/61, 50 Roosevelt Ave, 1050, Brussels, Belgium.
Present address: Institute of Mechanical, Process & Energy Engineering, Heriot-Watt University, Edinburgh, EH14 4AS, UK.
BMC Bioinformatics. 2017 Feb 10;18(1):95. doi: 10.1186/s12859-017-1528-9.
The identification of immunogenic regions on the surface of antigens, which are able to be recognized by antibodies and to trigger an immune response, is a major challenge for the design of new and effective vaccines. The prediction of such regions through computational immunology techniques is a challenging goal, which will ultimately lead to a drastic limitation of the experimental tests required to validate their efficiency. However, current methods are far from being sufficiently reliable and/or applicable on a large scale.
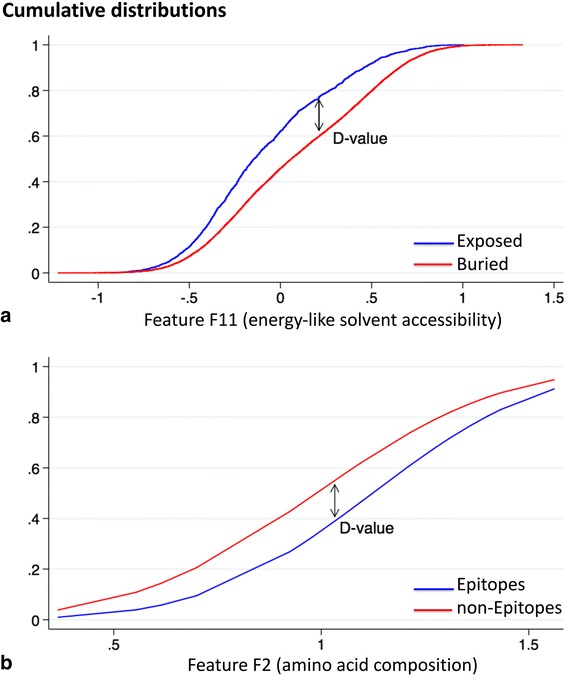
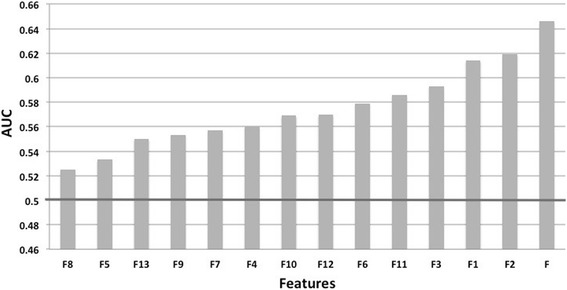
We developed SEPIa, a B-cell epitope predictor from the protein sequence, which is sufficiently fast to be applicable on a large scale. The originality of SEPIa lies in the combination of two classifiers, a naïve Bayesian and a random forest classifier, through a voting algorithm that exploits the advantages of both. It is based on 13 sequence-based features, whose values in a 9-residue sequence window are compiled to predict the epitope/non-epitope state of the central residue. The features are related to the type of amino acid, its conservation in homologous proteins, and its tendency of being exposed to the solvent, soluble, flexible, and disordered. The highest signal is obtained from statistical amino acid preferences, but all 13 features contribute non-negligibly in the predictor. SEPIa's average prediction accuracy is limited, with an AUC score (area under the receiver operating characteristic curve) that reaches 0.65 both in 10-fold cross-validation and on an independent test set. It is nevertheless slightly higher than that of other methods evaluated on the same test set.
SEPIa was applied to a test protein whose epitopes are known, human β2 adrenergic G-protein-coupled receptor, with promising results. Although the actual AUC score is rather low, many of the predicted epitopes cluster together and overlap the experimental epitope region. The reasons underlying the limitations of SEPIa and of all other B-cell epitope predictors are discussed.
识别抗原表面能够被抗体识别并引发免疫反应的免疫原性区域,是设计新型有效疫苗的一项重大挑战。通过计算免疫学技术预测此类区域是一个具有挑战性的目标,这最终将极大地限制验证其有效性所需的实验测试。然而,目前的方法远未达到足够可靠和/或可大规模应用的程度。
我们开发了SEPIa,一种基于蛋白质序列的B细胞表位预测器,其速度足够快,可大规模应用。SEPIa的独特之处在于通过一种利用两者优势的投票算法,将朴素贝叶斯和随机森林分类器这两种分类器相结合。它基于13个基于序列的特征,这些特征在一个9残基序列窗口中的值被汇总起来,以预测中心残基的表位/非表位状态。这些特征与氨基酸类型、其在同源蛋白中的保守性以及其暴露于溶剂、可溶、灵活和无序的倾向有关。最高信号来自统计氨基酸偏好,但所有13个特征在预测器中都有不可忽视的贡献。SEPIa的平均预测准确率有限,在10折交叉验证和独立测试集上,其AUC分数(受试者工作特征曲线下面积)均达到0.65。不过,它仍略高于在同一测试集上评估的其他方法。
SEPIa被应用于一种已知表位的测试蛋白,即人β2肾上腺素能G蛋白偶联受体,结果令人鼓舞。尽管实际的AUC分数相当低,但许多预测的表位聚集在一起并与实验表位区域重叠。讨论了SEPIa和所有其他B细胞表位预测器局限性的潜在原因。