Kashaf Sara Saheb, Angione Claudio, Lió Pietro
Computer Laboratory, University of Cambridge, 15 JJ Thomson Avenue, Cambridge, CB3 0FD, UK.
Department of Computer Science and Information Systems, Teesside University, Borough road, Middlesbrough, TS1 3BA, UK.
BMC Syst Biol. 2017 Feb 16;11(1):25. doi: 10.1186/s12918-017-0395-3.
Clostridium difficile is a bacterium which can infect various animal species, including humans. Infection with this bacterium is a leading healthcare-associated illness. A better understanding of this organism and the relationship between its genotype and phenotype is essential to the search for an effective treatment. Genome-scale metabolic models contain all known biochemical reactions of a microorganism and can be used to investigate this relationship.
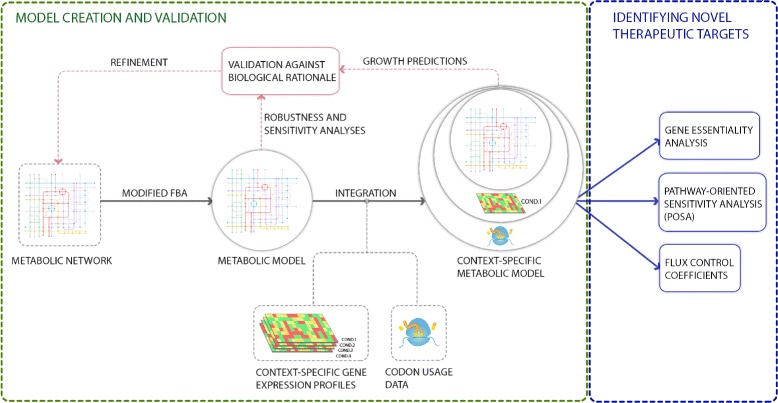
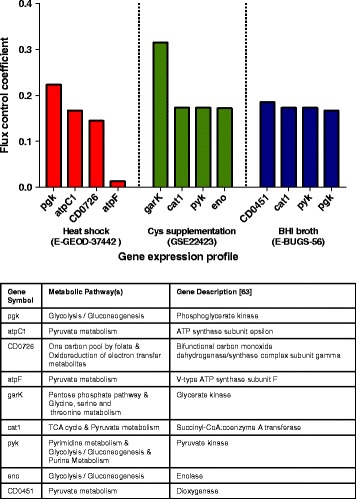
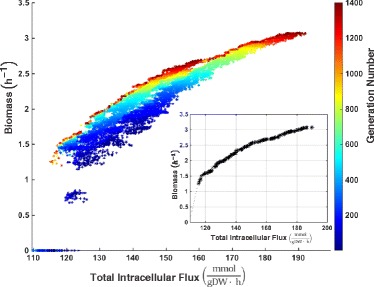
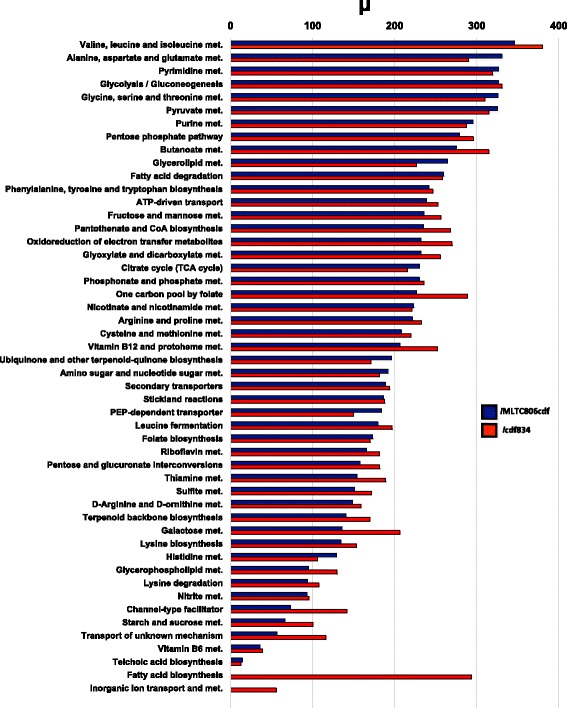
We present icdf834, an updated metabolic network of C. difficile that builds on iMLTC806cdf and features 1227 reactions, 834 genes, and 807 metabolites. We used this metabolic network to reconstruct the metabolic landscape of this bacterium. The standard metabolic model cannot account for changes in the bacterial metabolism in response to different environmental conditions. To account for this limitation, we also integrated transcriptomic data, which details the gene expression of the bacterium in a wide array of environments. Importantly, to bridge the gap between gene expression levels and protein abundance, we accounted for the synonymous codon usage bias of the bacterium in the model. To our knowledge, this is the first time codon usage has been quantified and integrated into a metabolic model. The metabolic fluxes were defined as a function of protein abundance. To determine potential therapeutic targets using the model, we conducted gene essentiality and metabolic pathway sensitivity analyses and calculated flux control coefficients. We obtained 92.3% accuracy in predicting gene essentiality when compared to experimental data for C. difficile R20291 (ribotype 027) homologs. We validated our context-specific metabolic models using sensitivity and robustness analyses and compared model predictions with literature on C. difficile. The model predicts interesting facets of the bacterium's metabolism, such as changes in the bacterium's growth in response to different environmental conditions.
After an extensive validation process, we used icdf834 to obtain state-of-the-art predictions of therapeutic targets for C. difficile. We show how context-specific metabolic models augmented with codon usage information can be a beneficial resource for better understanding C. difficile and for identifying novel therapeutic targets. We remark that our approach can be applied to investigate and treat against other pathogens.
艰难梭菌是一种可感染包括人类在内的多种动物物种的细菌。该细菌感染是一种主要的医疗保健相关疾病。更好地了解这种微生物及其基因型与表型之间的关系对于寻找有效的治疗方法至关重要。基因组规模代谢模型包含微生物所有已知的生化反应,可用于研究这种关系。
我们展示了icdf834,这是一种基于iMLTC806cdf更新的艰难梭菌代谢网络,具有1227个反应、834个基因和807个代谢物。我们使用这个代谢网络重建了该细菌的代谢格局。标准代谢模型无法解释细菌代谢对不同环境条件的变化。为了克服这一局限性,我们还整合了转录组数据,该数据详细说明了该细菌在多种环境中的基因表达。重要的是,为了弥合基因表达水平与蛋白质丰度之间的差距,我们在模型中考虑了该细菌的同义密码子使用偏好。据我们所知,这是首次对密码子使用进行量化并将其整合到代谢模型中。代谢通量被定义为蛋白质丰度的函数。为了使用该模型确定潜在的治疗靶点,我们进行了基因必需性和代谢途径敏感性分析,并计算了通量控制系数。与艰难梭菌R20291(核糖体分型027)同源物的实验数据相比,我们在预测基因必需性方面获得了92.3%的准确率。我们使用敏感性和稳健性分析验证了我们的上下文特异性代谢模型,并将模型预测与关于艰难梭菌的文献进行了比较。该模型预测了该细菌代谢的有趣方面,例如该细菌在不同环境条件下生长的变化。
经过广泛的验证过程后,我们使用icdf834获得了艰难梭菌治疗靶点的最新预测。我们展示了如何通过密码子使用信息增强上下文特异性代谢模型,这对于更好地了解艰难梭菌和识别新的治疗靶点可能是一个有益的资源。我们指出,我们的方法可应用于研究和对抗其他病原体。