Rappaport Lab, UC Berkeley, School of Public Health , GL81 Koshland Hall, Berkeley, California 94720, United States.
Metabolomics FiehnLab, NIH West-Coast Metabolomics Center (WCMC), University of California Davis , Davis, California 95616 United States.
Anal Chem. 2017 Apr 4;89(7):3919-3928. doi: 10.1021/acs.analchem.6b02394. Epub 2017 Mar 27.
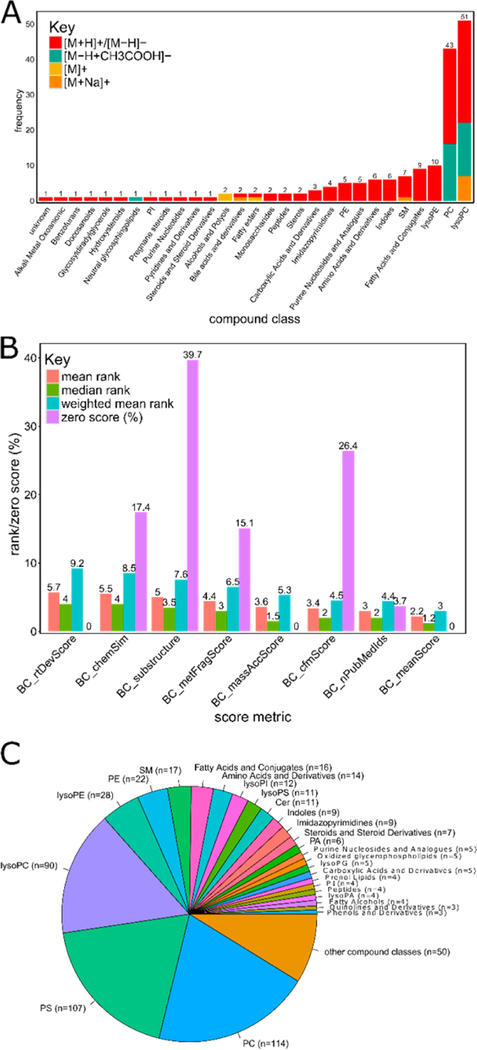
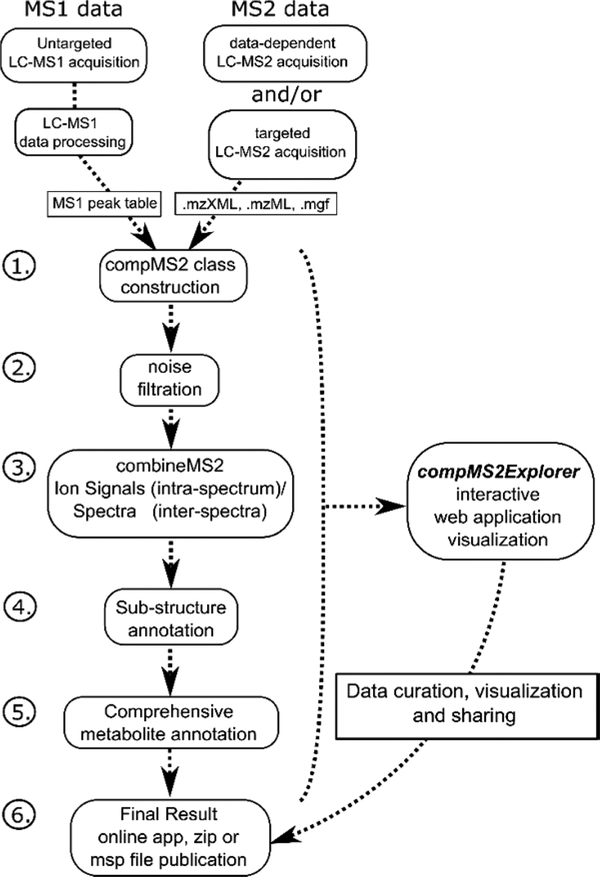
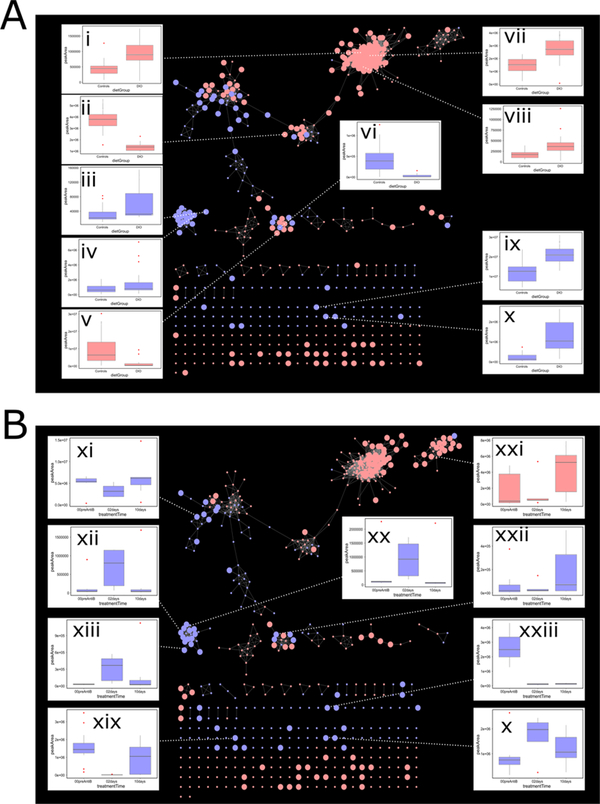
A long-standing challenge of untargeted metabolomic profiling by ultrahigh-performance liquid chromatography-high-resolution mass spectrometry (UHPLC-HRMS) is efficient transition from unknown mass spectral features to confident metabolite annotations. The compMSMiner (Comprehensive MS Miner) package was developed in the R language to facilitate rapid, comprehensive feature annotation using a peak-picker-output and MS data files as inputs. The number of MS spectra that can be collected during a metabolomic profiling experiment far outweigh the amount of time required for pain-staking manual interpretation; therefore, a degree of software workflow autonomy is required for broad-scale metabolite annotation. CompMSMiner integrates many useful tools in a single workflow for metabolite annotation and also provides a means to overview the MS data with a Web application GUI compMSExplorer (Comprehensive MS Explorer) that also facilitates data-sharing and transparency. The automatable compMSMiner workflow consists of the following steps: (i) matching unknown MS features to precursor MS scans, (ii) filtration of spectral noise (dynamic noise filter), (iii) generation of composite mass spectra by multiple similar spectrum signal summation and redundant/contaminant spectra removal, (iv) interpretation of possible fragment ion substructure using an internal database, (v) annotation of unknowns with chemical and spectral databases with prediction of mammalian biotransformation metabolites, wrapper functions for in silico fragmentation software, nearest neighbor chemical similarity scoring, random forest based retention time prediction, text-mining based false positive removal/true positive ranking, chemical taxonomic prediction and differential evolution based global annotation score optimization, and (vi) network graph visualizations, data curation, and sharing are made possible via the compMSExplorer application. Metabolite identities and comments can also be recorded using an interactive table within compMSExplorer. The utility of the package is illustrated with a data set of blood serum samples from 7 diet induced obese (DIO) and 7 nonobese (NO) C57BL/6J mice, which were also treated with an antibiotic (streptomycin) to knockdown the gut microbiota. The results of fully autonomous and objective usage of compMSMiner are presented here. All automatically annotated spectra output by the workflow are provided in the Supporting Information and can alternatively be explored as publically available compMSExplorer applications for both positive and negative modes ( https://wmbedmands.shinyapps.io/compMS2_mouseSera_POS and https://wmbedmands.shinyapps.io/compMS2_mouseSera_NEG ). The workflow provided rapid annotation of a diversity of endogenous and gut microbially derived metabolites affected by both diet and antibiotic treatment, which conformed to previously published reports. Composite spectra (n = 173) were autonomously matched to entries of the Massbank of North America (MoNA) spectral repository. These experimental and virtual (lipidBlast) spectra corresponded to 29 common endogenous compound classes (e.g., 51 lysophosphatidylcholines spectra) and were then used to calculate the ranking capability of 7 individual scoring metrics. It was found that an average of the 7 individual scoring metrics provided the most effective weighted average ranking ability of 3 for the MoNA matched spectra in spite of potential risk of false positive annotations emerging from automation. Minor structural differences such as relative carbon-carbon double bond positions were found in several cases to affect the correct rank of the MoNA annotated metabolite. The latest release and an example workflow is available in the package vignette ( https://github.com/WMBEdmands/compMS2Miner ) and a version of the published application is available on the shinyapps.io site ( https://wmbedmands.shinyapps.io/compMS2Example ).
长期以来,超高效液相色谱-高分辨质谱(UHPLC-HRMS)非靶向代谢组学分析的一个挑战是如何将未知的质谱特征有效地转换为可靠的代谢物注释。Comprehensive MS Miner (CompMSMiner) 包是用 R 语言开发的,用于在输入峰提取器输出和 MS 数据文件的情况下,快速、全面地进行特征注释。在代谢组学分析实验中,MS 谱的数量远远超过手动解释所需的时间;因此,需要一定程度的软件工作流程自动化来进行广泛的代谢物注释。CompMSMiner 将许多有用的工具集成到一个工作流程中,用于代谢物注释,同时还提供了一个使用 Web 应用程序 GUI compMSExplorer (Comprehensive MS Explorer)的 MS 数据概览方式,该应用程序还便于数据共享和透明度。自动化的 CompMSMiner 工作流程包括以下步骤:(i)将未知的 MS 特征与前体 MS 扫描进行匹配,(ii)过滤光谱噪声(动态噪声滤波器),(iii)通过多个相似谱信号的总和和冗余/污染物谱的去除生成复合质谱,(iv)使用内部数据库解释可能的碎片离子亚结构,(v)使用化学和光谱数据库对未知物进行注释,并预测哺乳动物生物转化代谢物,用于计算的片段化软件的包装函数,基于最近邻化学相似性评分的保留时间预测,基于随机森林的保留时间预测,基于文本挖掘的假阳性去除/真阳性排序,化学分类预测和基于差异进化的全局注释评分优化,以及(vi)通过 compMSExplorer 应用程序实现网络图形可视化、数据管理和共享。也可以使用 compMSExplorer 中的交互式表格记录代谢物的身份和注释。本文通过一组来自 7 只饮食诱导肥胖(DIO)和 7 只非肥胖(NO)C57BL/6J 小鼠的血清样本数据,以及一组用抗生素(链霉素)处理以敲低肠道微生物群的血清样本数据,说明了该软件包的实用性。工作流程中自动注释的所有 MS 谱都在支持信息中提供,也可以作为公共的 compMSExplorer 应用程序(分别用于正模式和负模式)进行探索(https://wmbedmands.shinyapps.io/compMS2_mouseSera_POS 和 https://wmbedmands.shinyapps.io/compMS2_mouseSera_NEG)。该工作流程提供了对受饮食和抗生素处理影响的多种内源性和肠道微生物衍生代谢物的快速注释,与之前发表的报告一致。(n = 173)的复合谱自动匹配到北美质谱库(MoNA)光谱库中的条目。这些实验和虚拟(脂质 Blast)谱对应于 29 种常见的内源性化合物类(例如,51 种溶血磷脂酰胆碱谱),然后用于计算 7 种单个评分指标的排名能力。结果发现,尽管自动化可能会出现假阳性注释的风险,但 7 种单个评分指标的平均值提供了对 MoNA 匹配谱的最有效的加权平均排名能力 3。在几个情况下,发现相对碳-碳双键位置等细微结构差异会影响 MoNA 注释代谢物的正确排名。最新版本和示例工作流程可在包说明(https://github.com/WMBEdmands/compMS2Miner)中获得,已发布应用程序的版本可在 shinyapps.io 网站上获得(https://wmbedmands.shinyapps.io/compMS2Example)。