Department of Chemical Engineering, Universitat Rovira i Virgili, Tarragona, Spain.
Department of Electronic Engineering, Metabolomics Platform, IISPV, Universitat Rovira i Virgili, Tarragona, Spain.
Bioinformatics. 2019 Oct 15;35(20):4089-4097. doi: 10.1093/bioinformatics/btz207.
The analysis of biological samples in untargeted metabolomic studies using LC-MS yields tens of thousands of ion signals. Annotating these features is of the utmost importance for answering questions as fundamental as, e.g. how many metabolites are there in a given sample.
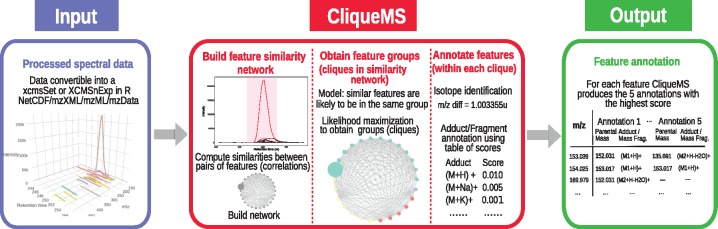
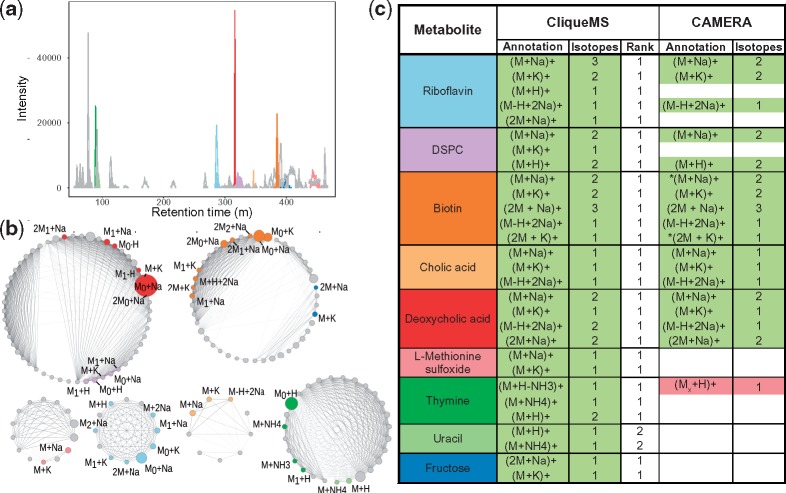
Here, we introduce CliqueMS, a new algorithm for annotating in-source LC-MS1 data. CliqueMS is based on the similarity between coelution profiles and therefore, as opposed to most methods, allows for the annotation of a single spectrum. Furthermore, CliqueMS improves upon the state of the art in several dimensions: (i) it uses a more discriminatory feature similarity metric; (ii) it treats the similarities between features in a transparent way by means of a simple generative model; (iii) it uses a well-grounded maximum likelihood inference approach to group features; (iv) it uses empirical adduct frequencies to identify the parental mass and (v) it deals more flexibly with the identification of the parental mass by proposing and ranking alternative annotations. We validate our approach with simple mixtures of standards and with real complex biological samples. CliqueMS reduces the thousands of features typically obtained in complex samples to hundreds of metabolites, and it is able to correctly annotate more metabolites and adducts from a single spectrum than available tools.
https://CRAN.R-project.org/package=cliqueMS and https://github.com/osenan/cliqueMS.
Supplementary data are available at Bioinformatics online.
在使用 LC-MS 的非靶向代谢组学研究中分析生物样本会产生成千上万的离子信号。对这些特征进行注释对于回答基本问题至关重要,例如,给定样本中有多少种代谢物。
在这里,我们引入了 CliqueMS,这是一种用于注释源内 LC-MS1 数据的新算法。CliqueMS 基于共洗脱曲线之间的相似性,因此与大多数方法不同,它允许注释单个光谱。此外,CliqueMS 在几个方面优于现有技术:(i)它使用更具区分性的特征相似性度量;(ii)它通过简单的生成模型以透明的方式处理特征之间的相似性;(iii)它使用基于充分理由的最大似然推断方法来对特征进行分组;(iv)它使用经验性加合物频率来识别母体质量;(v)它通过提出和排序替代注释来更灵活地处理母体质量的识别。我们使用简单的标准混合物和真实的复杂生物样本验证了我们的方法。CliqueMS 将复杂样本中通常获得的数千个特征减少到数百个代谢物,并且它能够从单个光谱中正确注释更多的代谢物和加合物,而不是可用的工具。
https://CRAN.R-project.org/package=cliqueMS 和 https://github.com/osenan/cliqueMS。
补充数据可在 Bioinformatics 在线获取。