Michael G. DeGroote Institute for Infectious Disease Research, Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario L8N 3Z5, Canada.
Department of Molecular and Cellular Biology, University of Guelph, Guelph, Ontario N1G 2W1, Canada.
Nat Microbiol. 2017 Mar 6;2:17028. doi: 10.1038/nmicrobiol.2017.28.
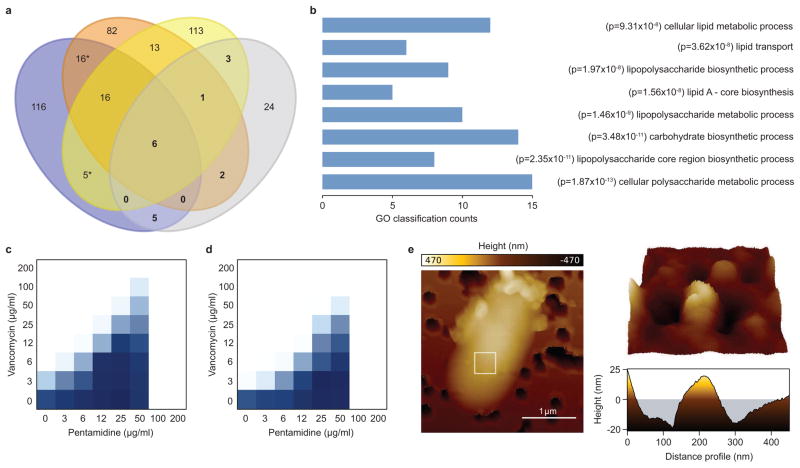
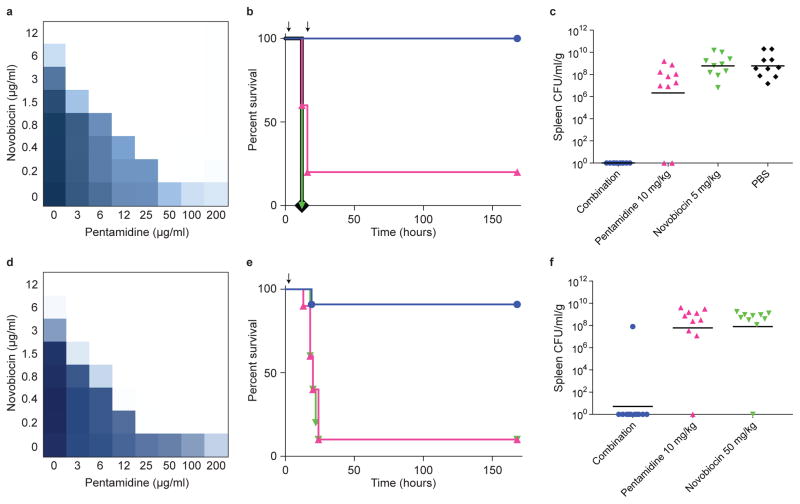
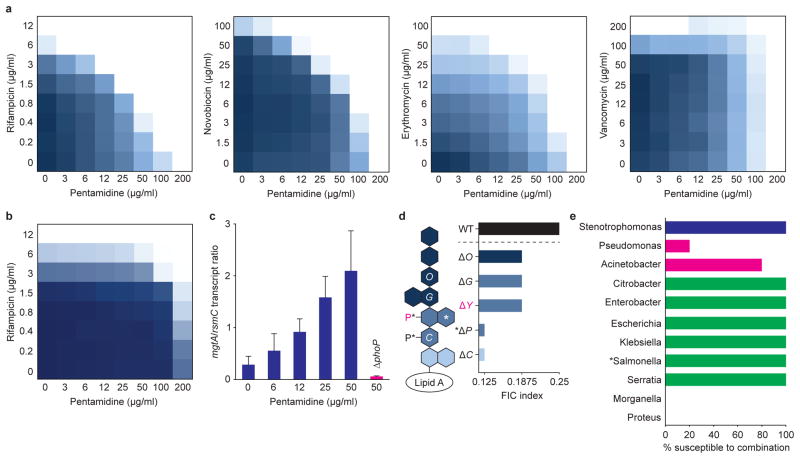
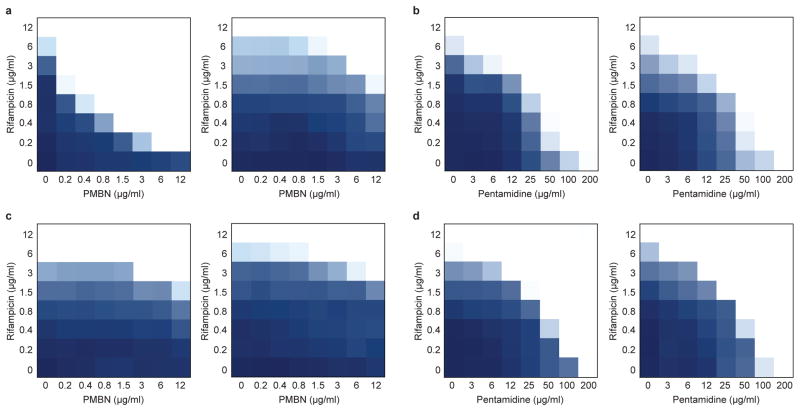
The increasing use of polymyxins in addition to the dissemination of plasmid-borne colistin resistance threatens to cause a serious breach in our last line of defence against multidrug-resistant Gram-negative pathogens, and heralds the emergence of truly pan-resistant infections. Colistin resistance often arises through covalent modification of lipid A with cationic residues such as phosphoethanolamine-as is mediated by Mcr-1 (ref. 2)-which reduce the affinity of polymyxins for lipopolysaccharide. Thus, new strategies are needed to address the rapidly diminishing number of treatment options for Gram-negative infections. The difficulty in eradicating Gram-negative bacteria is largely due to their highly impermeable outer membrane, which serves as a barrier to many otherwise effective antibiotics. Here, we describe an unconventional screening platform designed to enrich for non-lethal, outer-membrane-active compounds with potential as adjuvants for conventional antibiotics. This approach identified the antiprotozoal drug pentamidine as an effective perturbant of the Gram-negative outer membrane through its interaction with lipopolysaccharide. Pentamidine displayed synergy with antibiotics typically restricted to Gram-positive bacteria, yielding effective drug combinations with activity against a wide range of Gram-negative pathogens in vitro, and against systemic Acinetobacter baumannii infections in mice. Notably, the adjuvant activity of pentamidine persisted in polymyxin-resistant bacteria in vitro and in vivo. Overall, pentamidine and its structural analogues represent unexploited molecules for the treatment of Gram-negative infections, particularly those having acquired polymyxin resistance determinants.
越来越多的人将多黏菌素与质粒介导的黏菌素耐药性一起使用,这有可能严重破坏我们对抗多药耐药革兰氏阴性病原体的最后一道防线,并预示着真正的全耐药感染的出现。黏菌素耐药性通常通过带正电荷的残基(如磷酸乙醇胺)共价修饰脂质 A 而产生,这是由 Mcr-1(参考文献 2)介导的,从而降低了多黏菌素与脂多糖的亲和力。因此,需要新的策略来解决革兰氏阴性感染治疗方案迅速减少的问题。难以根除革兰氏阴性细菌主要是由于它们高度不透性的外膜,这是许多其他有效的抗生素的屏障。在这里,我们描述了一个非传统的筛选平台,旨在富集具有成为传统抗生素佐剂潜力的非致死性、外膜活性化合物。这种方法发现抗原生动物药物戊二脒通过与脂多糖相互作用成为革兰氏阴性外膜的有效扰动剂。戊二脒与通常仅限于革兰氏阳性细菌的抗生素表现出协同作用,在体外对广泛的革兰氏阴性病原体产生有效的药物组合,在体内对全身性鲍曼不动杆菌感染也有活性。值得注意的是,戊二脒在体外和体内的多黏菌素耐药菌中保持了佐剂活性。总的来说,戊二脒及其结构类似物代表了治疗革兰氏阴性感染的未开发分子,特别是那些已经获得多黏菌素耐药决定因素的感染。