Graham Elaina D, Heidelberg John F, Tully Benjamin J
Department of Biological Sciences, University of Southern California , Los Angeles , CA , USA.
Department of Biological Sciences, University of Southern California, Los Angeles, CA, USA; Center for Dark Energy Biosphere Investigations, Los Angeles, CA, USA.
PeerJ. 2017 Mar 8;5:e3035. doi: 10.7717/peerj.3035. eCollection 2017.
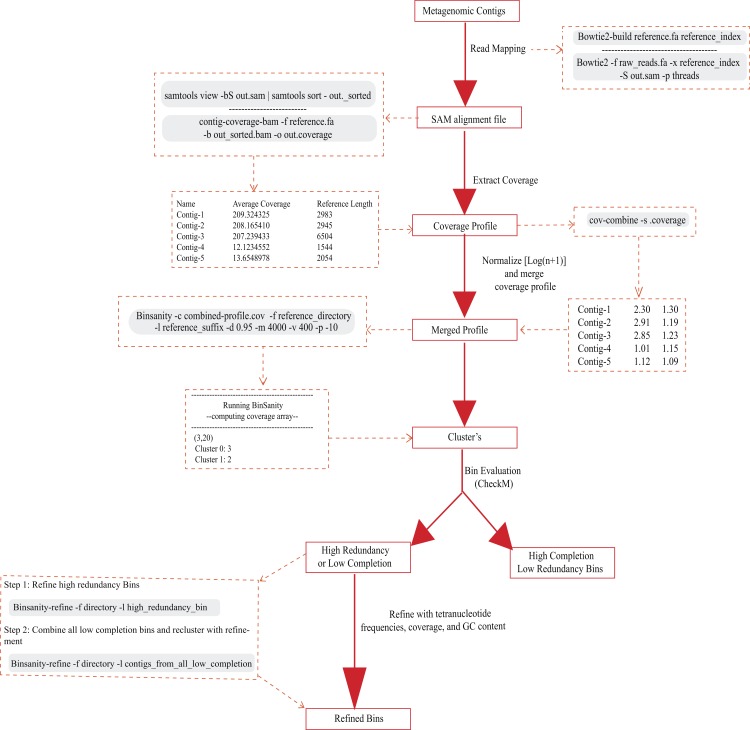
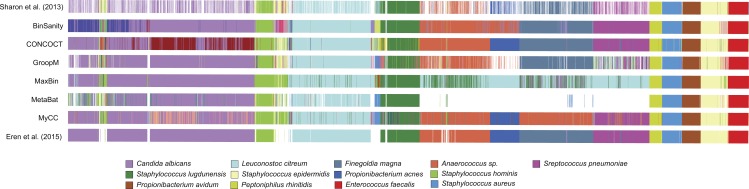
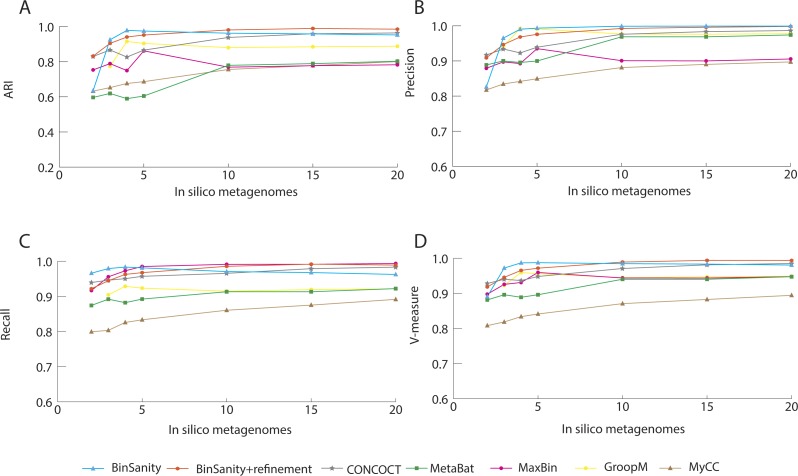
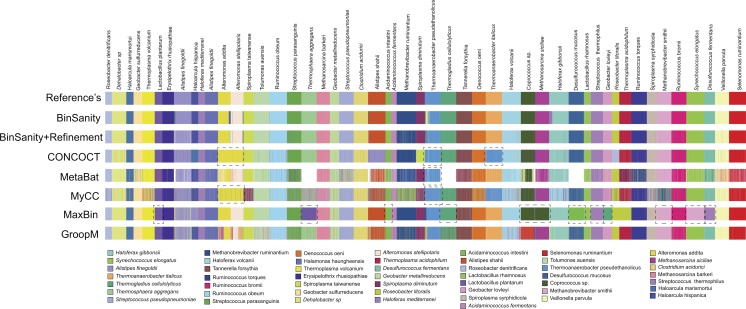
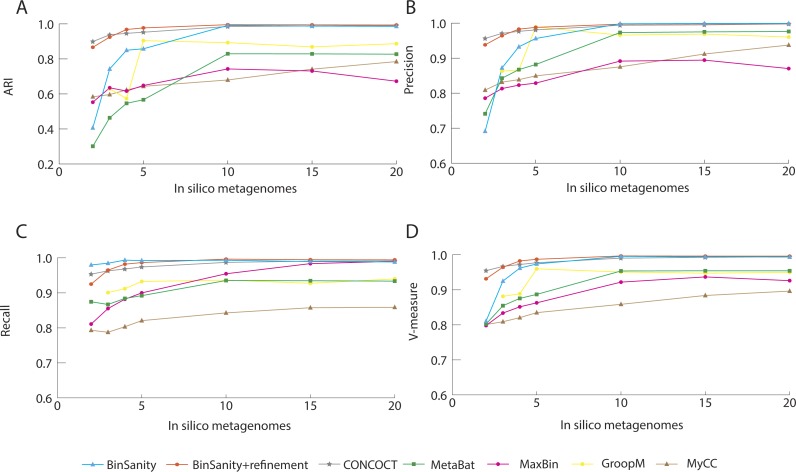
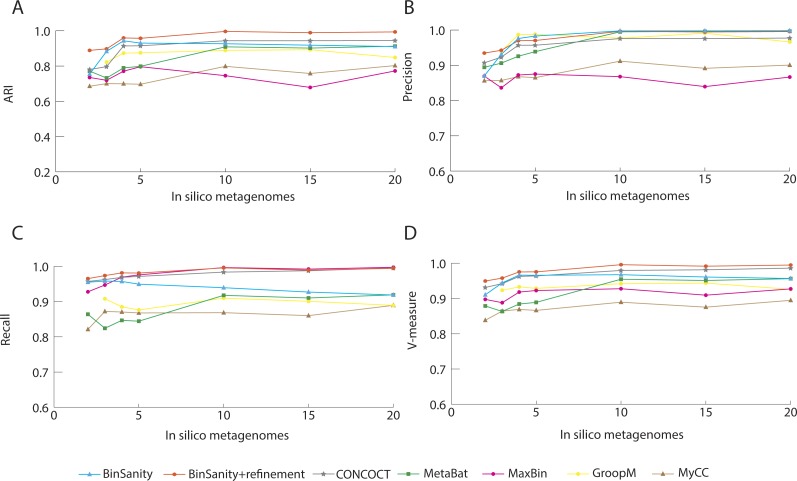
Metagenomics has become an integral part of defining microbial diversity in various environments. Many ecosystems have characteristically low biomass and few cultured representatives. Linking potential metabolisms to phylogeny in environmental microorganisms is important for interpreting microbial community functions and the impacts these communities have on geochemical cycles. However, with metagenomic studies there is the computational hurdle of 'binning' contigs into phylogenetically related units or putative genomes. Binning methods have been implemented with varying approaches such as k-means clustering, Gaussian mixture models, hierarchical clustering, neural networks, and two-way clustering; however, many of these suffer from biases against low coverage/abundance organisms and closely related taxa/strains. We are introducing a new binning method, BinSanity, that utilizes the clustering algorithm affinity propagation (AP), to cluster assemblies using coverage with compositional based refinement (tetranucleotide frequency and percent GC content) to optimize bins containing multiple source organisms. This separation of composition and coverage based clustering reduces bias for closely related taxa. BinSanity was developed and tested on artificial metagenomes varying in size and complexity. Results indicate that BinSanity has a higher precision, recall, and Adjusted Rand Index compared to five commonly implemented methods. When tested on a previously published environmental metagenome, BinSanity generated high completion and low redundancy bins corresponding with the published metagenome-assembled genomes.
宏基因组学已成为定义各种环境中微生物多样性不可或缺的一部分。许多生态系统具有典型的低生物量且培养出的代表性微生物很少。将环境微生物的潜在代谢与系统发育联系起来对于解释微生物群落功能以及这些群落对地球化学循环的影响至关重要。然而,在宏基因组学研究中,存在将重叠群“归类”到系统发育相关单元或假定基因组中的计算障碍。归类方法已通过多种不同方法实现,如k均值聚类、高斯混合模型、层次聚类、神经网络和双向聚类;然而,其中许多方法存在对低覆盖度/丰度生物以及密切相关的分类群/菌株的偏差。我们正在引入一种新的归类方法BinSanity,它利用聚类算法亲和传播(AP),使用覆盖度并结合基于组成的细化(四核苷酸频率和GC含量百分比)对组装序列进行聚类,以优化包含多种来源生物的分类单元。这种基于组成和覆盖度的聚类分离减少了对密切相关分类群的偏差。BinSanity是在大小和复杂度各异的人工宏基因组上开发和测试的。结果表明,与五种常用方法相比,BinSanity具有更高的精度、召回率和调整兰德指数。在先前发表的环境宏基因组上进行测试时,BinSanity生成了与已发表的宏基因组组装基因组相对应的高完整性和低冗余度的分类单元。