Wen Bo, Mei Zhanlong, Zeng Chunwei, Liu Siqi
BGI-Shenzhen, Shenzhen, 518083, China.
China National GeneBank-Shenzhen, BGI-Shenzhen, Shenzhen, Guangdong, 518083, China.
BMC Bioinformatics. 2017 Mar 21;18(1):183. doi: 10.1186/s12859-017-1579-y.
Non-targeted metabolomics based on mass spectrometry enables high-throughput profiling of the metabolites in a biological sample. The large amount of data generated from mass spectrometry requires intensive computational processing for annotation of mass spectra and identification of metabolites. Computational analysis tools that are fully integrated with multiple functions and are easily operated by users who lack extensive knowledge in programing are needed in this research field.
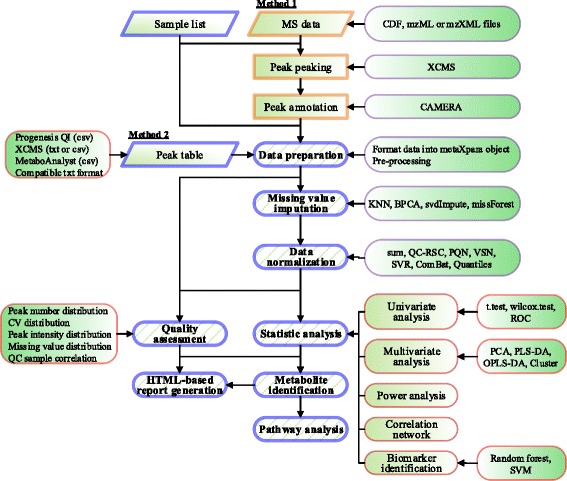
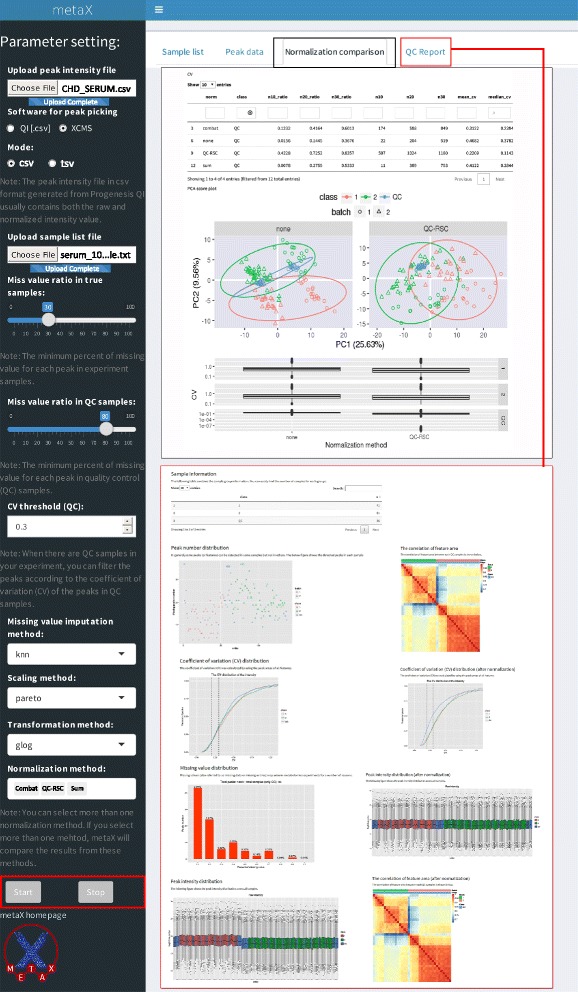
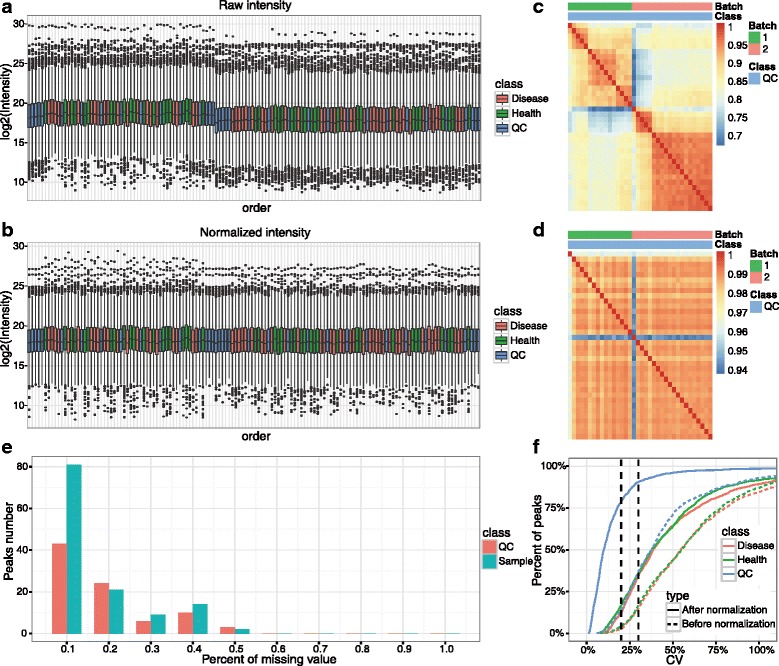
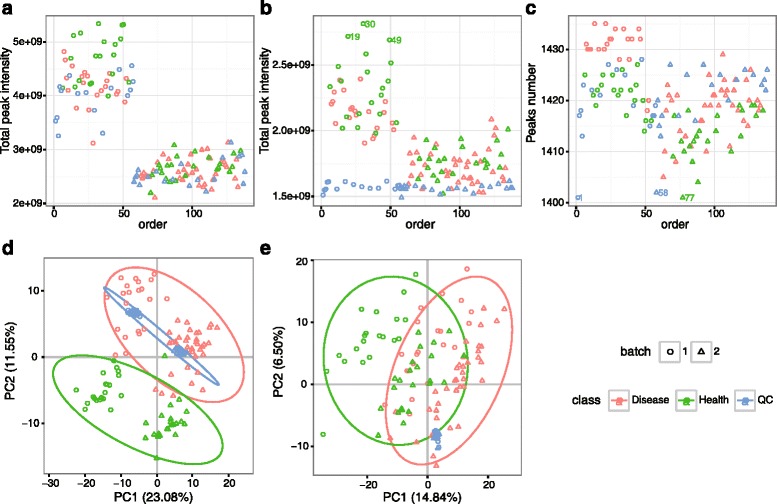
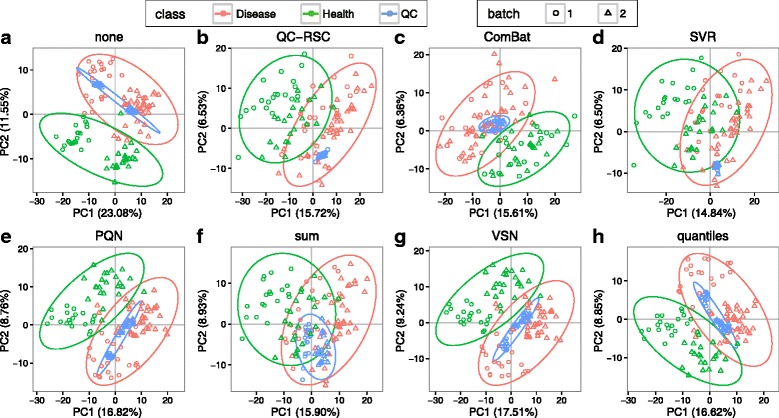
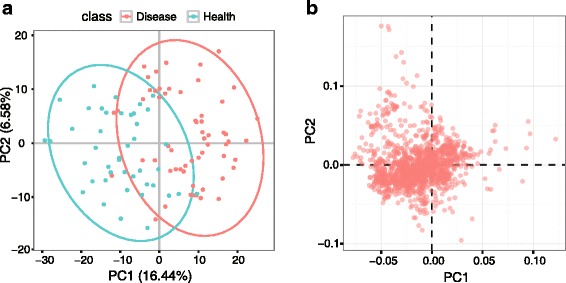
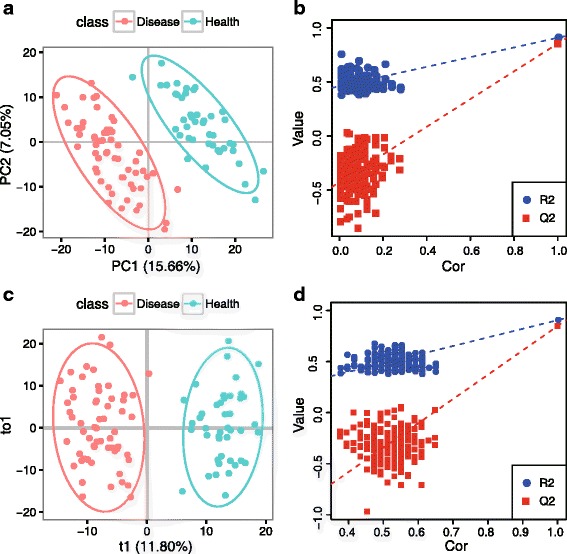
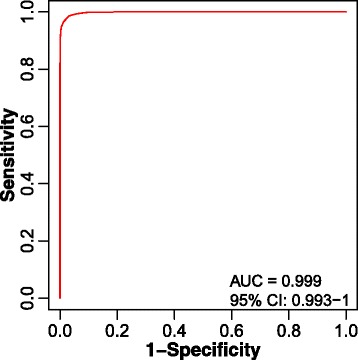
We herein developed an R package, metaX, that is capable of end-to-end metabolomics data analysis through a set of interchangeable modules. Specifically, metaX provides several functions, such as peak picking and annotation, data quality assessment, missing value imputation, data normalization, univariate and multivariate statistics, power analysis and sample size estimation, receiver operating characteristic analysis, biomarker selection, pathway annotation, correlation network analysis, and metabolite identification. In addition, metaX offers a web-based interface ( http://metax.genomics.cn ) for data quality assessment and normalization method evaluation, and it generates an HTML-based report with a visualized interface. The metaX utilities were demonstrated with a published metabolomics dataset on a large scale. The software is available for operation as either a web-based graphical user interface (GUI) or in the form of command line functions. The package and the example reports are available at http://metax.genomics.cn/ .
The pipeline of metaX is platform-independent and is easy to use for analysis of metabolomics data generated from mass spectrometry.
基于质谱的非靶向代谢组学能够对生物样品中的代谢物进行高通量分析。质谱产生的大量数据需要密集的计算处理,以用于质谱注释和代谢物鉴定。该研究领域需要与多种功能完全集成且易于缺乏广泛编程知识的用户操作的计算分析工具。
我们在此开发了一个R包metaX,它能够通过一组可互换的模块进行端到端的代谢组学数据分析。具体而言,metaX提供了多种功能,如峰提取与注释、数据质量评估、缺失值插补、数据归一化、单变量和多变量统计、功效分析和样本量估计、受试者工作特征分析、生物标志物选择、通路注释、相关网络分析以及代谢物鉴定。此外,metaX提供了一个基于网络的界面(http://metax.genomics.cn )用于数据质量评估和归一化方法评估,并生成具有可视化界面的基于HTML的报告。metaX工具在一个已发表的大规模代谢组学数据集上得到了验证。该软件既可以作为基于网络的图形用户界面(GUI)运行,也可以以命令行函数的形式运行。该软件包和示例报告可在http://metax.genomics.cn/ 获取。
metaX流程独立于平台,易于用于分析质谱产生的代谢组学数据。