Cheng Zhanzhan, Huang Kai, Wang Yang, Liu Hui, Guan Jihong, Zhou Shuigeng
School of Computer Science, Fudan University, Handan Road, Shanghai, 200433, China.
School of Computer Science, Jiangxi Normal University, Nanchang, 330022, China.
BMC Syst Biol. 2017 Mar 14;11(Suppl 2):9. doi: 10.1186/s12918-017-0390-8.
The identification of Protein-RNA Interactions (PRIs) is important to understanding cell activities. Recently, several machine learning-based methods have been developed for identifying PRIs. However, the performance of these methods is unsatisfactory. One major reason is that they usually use unreliable negative samples in the training process.
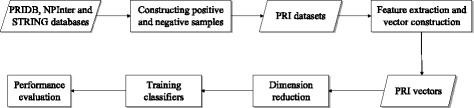
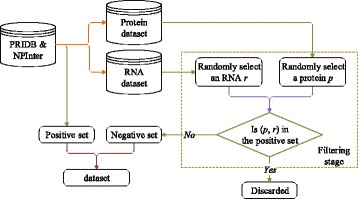
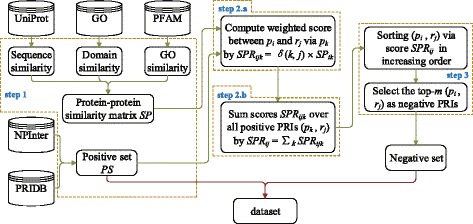
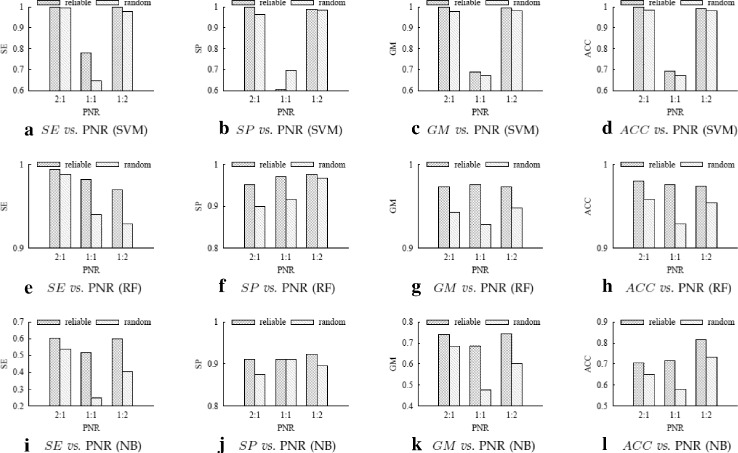
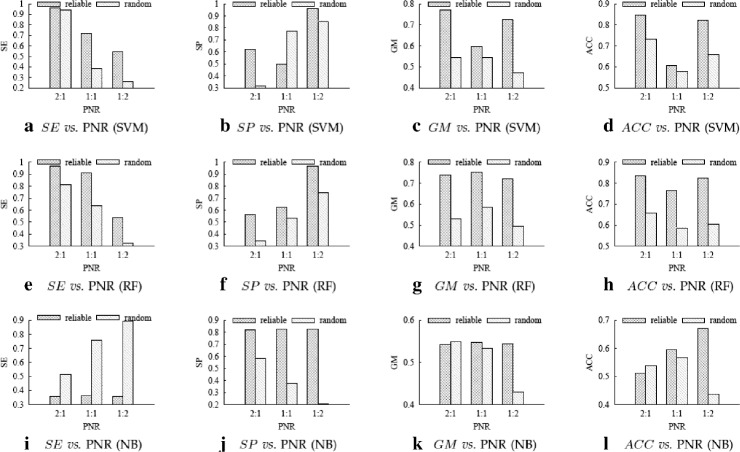
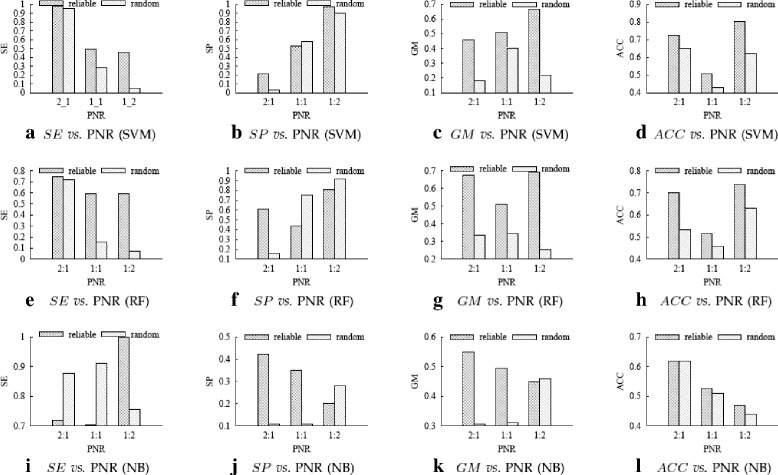
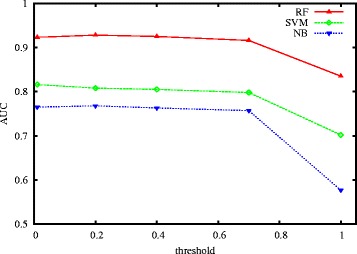
For boosting the performance of PRI prediction, we propose a novel method to generate reliable negative samples. Concretely, we firstly collect the known PRIs as positive samples for generating positive sets. For each positive set, we construct two corresponding negative sets, one is by our method and the other by random method. Each positive set is combined with a negative set to form a dataset for model training and performance evaluation. Consequently, we get 18 datasets of different species and different ratios of negative samples to positive samples. Secondly, sequence-based features are extracted to represent each of PRIs and protein-RNA pairs in the datasets. A filter-based method is employed to cut down the dimensionality of feature vectors for reducing computational cost. Finally, the performance of support vector machine (SVM), random forest (RF) and naive Bayes (NB) is evaluated on the generated 18 datasets.
Extensive experiments show that comparing to using randomly-generated negative samples, all classifiers achieve substantial performance improvement by using negative samples selected by our method. The improvements on accuracy and geometric mean for the SVM classifier, the RF classifier and the NB classifier are as high as 204.5 and 68.7%, 174.5 and 53.9%, 80.9 and 54.3%, respectively.
Our method is useful to the identification of PRIs.
蛋白质 - RNA 相互作用(PRIs)的识别对于理解细胞活动至关重要。最近,已经开发了几种基于机器学习的方法来识别 PRIs。然而,这些方法的性能并不理想。一个主要原因是它们在训练过程中通常使用不可靠的负样本。
为了提高 PRI 预测的性能,我们提出了一种生成可靠负样本的新方法。具体来说,我们首先收集已知的 PRIs 作为正样本以生成正集。对于每个正集,我们构建两个相应的负集,一个通过我们的方法,另一个通过随机方法。每个正集与一个负集组合形成一个用于模型训练和性能评估的数据集。因此,我们得到了 18 个不同物种以及负样本与正样本不同比例的数据集。其次,提取基于序列的特征来表示数据集中的每个 PRIs 和蛋白质 - RNA 对。采用基于滤波器的方法来降低特征向量的维度以降低计算成本。最后,在生成的 18 个数据集上评估支持向量机(SVM)、随机森林(RF)和朴素贝叶斯(NB)的性能。
大量实验表明,与使用随机生成的负样本相比,所有分类器通过使用我们方法选择的负样本都实现了显著的性能提升。SVM 分类器、RF 分类器和 NB 分类器在准确率和几何平均值上的提升分别高达 204.5%和 68.7%、174.5%和 53.9%、80.9%和 54.3%。
我们的方法对 PRIs 的识别很有用。