Sahadeo N S D, Allicock O M, De Salazar P M, Auguste A J, Widen S, Olowokure B, Gutierrez C, Valadere A M, Polson-Edwards K, Weaver S C, Carrington C V F
Department of Preclinical Sciences, Faculty of Medical Sciences, The University of the West Indies, St. Augustine Campus, Trinidad and Tobago.
Caribbean Public Health Agency, Port-of-Spain, Trinidad and Tobago.
Virus Evol. 2017 May 3;3(1):vex010. doi: 10.1093/ve/vex010. eCollection 2017 Jan.
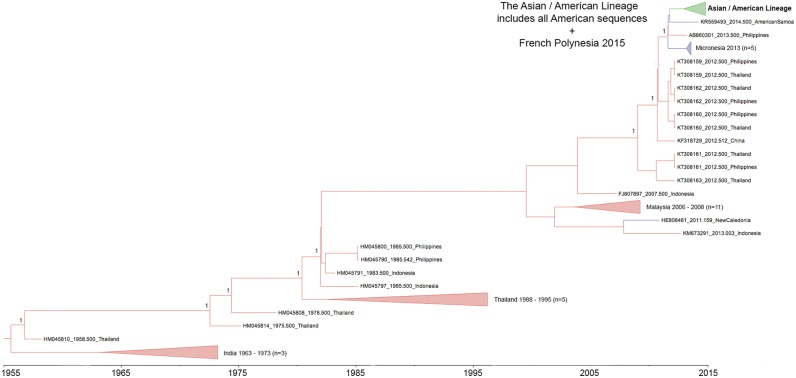
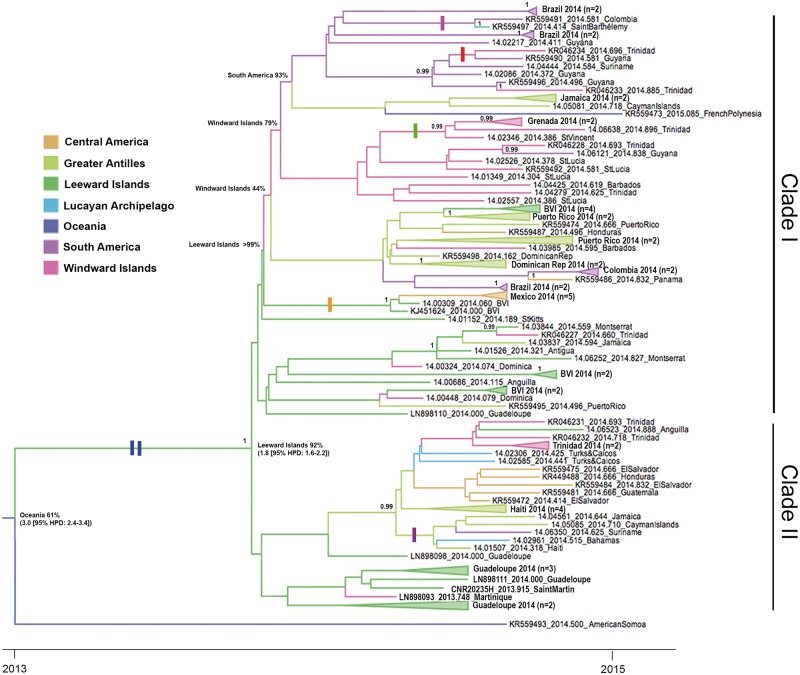
Local transmission of chikungunya virus (CHIKV) was first detected in the Americas in December 2013, after which it spread rapidly throughout the Caribbean islands and American mainland, causing a major chikungunya fever epidemic. Previous phylogenetic analysis of CHIKV from a limited number of countries in the Americas suggests that an Asian genotype strain was responsible, except in Brazil where both Asian and East/Central/South African (ECSA) lineage strains were detected. In this study, we sequenced thirty-three complete CHIKV genomes from viruses isolated in 2014 from fourteen Caribbean islands, the Bahamas and two mainland countries in the Americas. Phylogenetic analyses confirmed that they all belonged to the Asian genotype and clustered together with other Caribbean and mainland sequences isolated during the American outbreak, forming an 'Asian/American' lineage defined by two amino acid substitutions, E2 V368A and 6K L20M, and divided into two well-supported clades. This lineage is estimated to be evolving at a mean rate of 5 × 10 substitutions per site per year (95% higher probability density, 2.9-7.9 × 10) and to have arisen from an ancestor introduced to the Caribbean (most likely from Oceania) in about March 2013, 9 months prior to the first report of CHIKV in the Americas. Estimation of evolutionary rates for individual gene regions and selection analyses indicate that (in contrast to the Indian Ocean Lineage that emerged from the ECSA genotype followed by adaptive evolution and with a significantly higher substitution rate) the evolutionary dynamics of the Asian/American lineage are very similar to the rest of the Asian genotype and natural selection does not appear to have played a major role in its emergence. However, several codon sites with evidence of positive selection were identified within the non-structural regions of Asian genotype sequences outside of the Asian/American lineage.
2013年12月,美洲首次检测到基孔肯雅病毒(CHIKV)的本地传播,此后该病毒迅速蔓延至整个加勒比群岛和美洲大陆,引发了一场严重的基孔肯雅热疫情。此前对来自美洲少数国家的CHIKV进行的系统发育分析表明,除巴西检测到亚洲基因型毒株和东非/中非/南非(ECSA)谱系毒株外,疫情由亚洲基因型毒株引起。在本研究中,我们对2014年从14个加勒比岛屿、巴哈马群岛和美洲两个大陆国家分离出的病毒的33个完整CHIKV基因组进行了测序。系统发育分析证实,它们均属于亚洲基因型,与美洲疫情期间分离出的其他加勒比和大陆序列聚集在一起,形成了一个由两个氨基酸替换(E2 V368A和6K L20M)定义的“亚洲/美洲”谱系,并分为两个得到充分支持的进化枝。据估计,该谱系的进化平均速率为每年每个位点5×10个替换(95%较高概率密度,2.9 - 7.9×10),大约在2013年3月(即美洲首次报告CHIKV的9个月前)由引入加勒比地区(很可能来自大洋洲)的一个祖先演化而来。对各个基因区域的进化速率估计和选择分析表明,(与从ECSA基因型出现并随后经历适应性进化且替换率显著更高的印度洋谱系不同)亚洲/美洲谱系的进化动态与亚洲基因型的其他部分非常相似,自然选择在其出现过程中似乎未起主要作用。然而,在亚洲/美洲谱系之外的亚洲基因型序列的非结构区域内,鉴定出了几个有正选择证据的密码子位点。