Rasigraf Olivia, Schmitt Julia, Jetten Mike S M, Lüke Claudia
Department of Microbiology, IWWR, Radboud University Nijmegen, Nijmegen, Netherlands.
DVGW-Forschungsstelle TUHH, Hamburg University of Technology, Hamburg, Germany.
Microbiologyopen. 2017 Aug;6(4). doi: 10.1002/mbo3.475. Epub 2017 May 23.
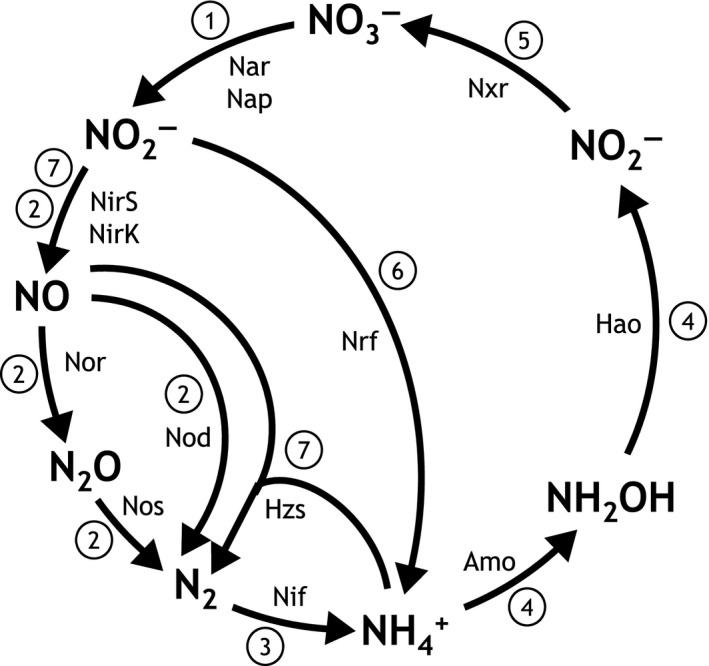
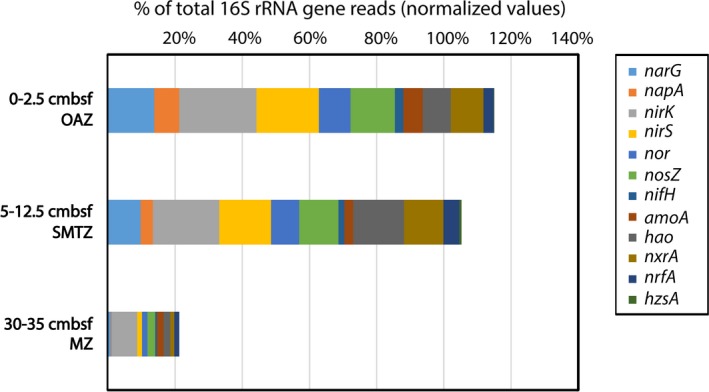
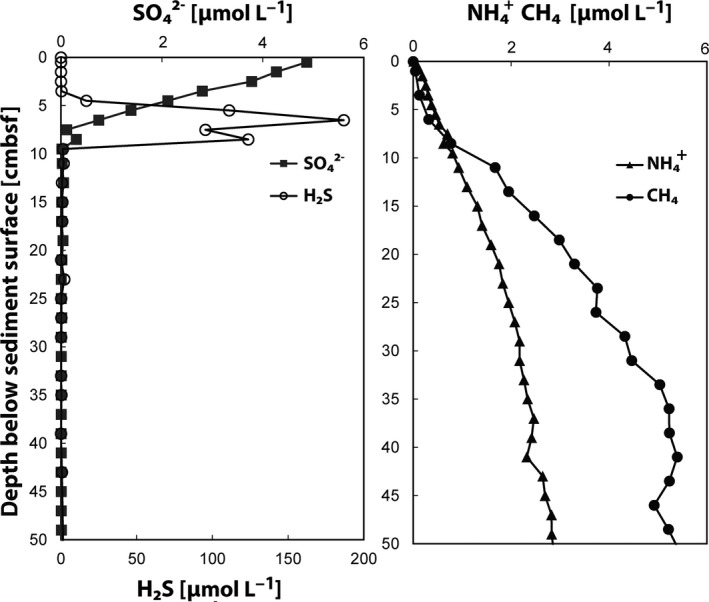
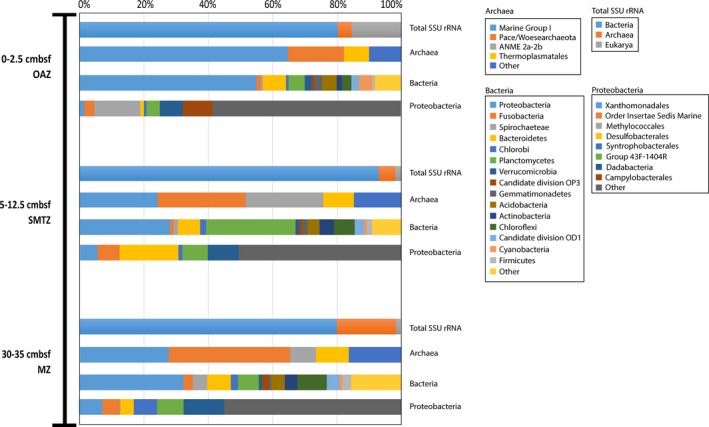
The biological nitrogen cycle is driven by a plethora of reactions transforming nitrogen compounds between various redox states. Here, we investigated the metagenomic potential for nitrogen cycle of the in situ microbial community in an oligotrophic, brackish environment of the Bothnian Sea sediment. Total DNA from three sediment depths was isolated and sequenced. The characterization of the total community was performed based on 16S rRNA gene inventory using SILVA database as reference. The diversity of diagnostic functional genes coding for nitrate reductases (napA;narG), nitrite:nitrate oxidoreductase (nxrA), nitrite reductases (nirK;nirS;nrfA), nitric oxide reductase (nor), nitrous oxide reductase (nosZ), hydrazine synthase (hzsA), ammonia monooxygenase (amoA), hydroxylamine oxidoreductase (hao), and nitrogenase (nifH) was analyzed by blastx against curated reference databases. In addition, Polymerase chain reaction (PCR)-based amplification was performed on the hzsA gene of anammox bacteria. Our results reveal high genomic potential for full denitrification to N , but minor importance of anaerobic ammonium oxidation and dissimilatory nitrite reduction to ammonium. Genomic potential for aerobic ammonia oxidation was dominated by Thaumarchaeota. A higher diversity of anammox bacteria was detected in metagenomes than with PCR-based technique. The results reveal the importance of various N-cycle driving processes and highlight the advantage of metagenomics in detection of novel microbial key players.
生物氮循环由大量反应驱动,这些反应在各种氧化还原状态之间转化含氮化合物。在此,我们研究了波罗的海贫营养微咸环境沉积物中原位微生物群落氮循环的宏基因组潜力。从三个沉积物深度分离出总DNA并进行测序。以SILVA数据库为参考,基于16S rRNA基因清单对整个群落进行表征。通过blastx对经过整理的参考数据库分析编码硝酸还原酶(napA;narG)、亚硝酸:硝酸氧化还原酶(nxrA)、亚硝酸还原酶(nirK;nirS;nrfA)、一氧化氮还原酶(nor)、氧化亚氮还原酶(nosZ)、肼合酶(hzsA)、氨单加氧酶(amoA)、羟胺氧化还原酶(hao)和固氮酶(nifH)的诊断功能基因的多样性。此外,对厌氧氨氧化细菌的hzsA基因进行基于聚合酶链反应(PCR)的扩增。我们的结果表明,将氮完全反硝化的基因组潜力很高,但厌氧氨氧化和异化亚硝酸盐还原为铵的重要性较小。需氧氨氧化的基因组潜力以奇古菌为主。在宏基因组中检测到的厌氧氨氧化细菌的多样性高于基于PCR的技术。结果揭示了各种氮循环驱动过程的重要性,并突出了宏基因组学在检测新型微生物关键参与者方面的优势。