de Torrente Laurence, Zimmerman Samuel, Taylor Deanne, Hasegawa Yu, Wells Christine A, Mar Jessica C
Department of Systems and Computational Biology, Albert Einstein College of Medicine, Bronx, NY, United States of America.
Department of Biomedical and Health Informatics, Children's Hospital of Philadelphia, Philadelphia, PA, United States of America.
PeerJ. 2017 May 23;5:e3334. doi: 10.7717/peerj.3334. eCollection 2017.
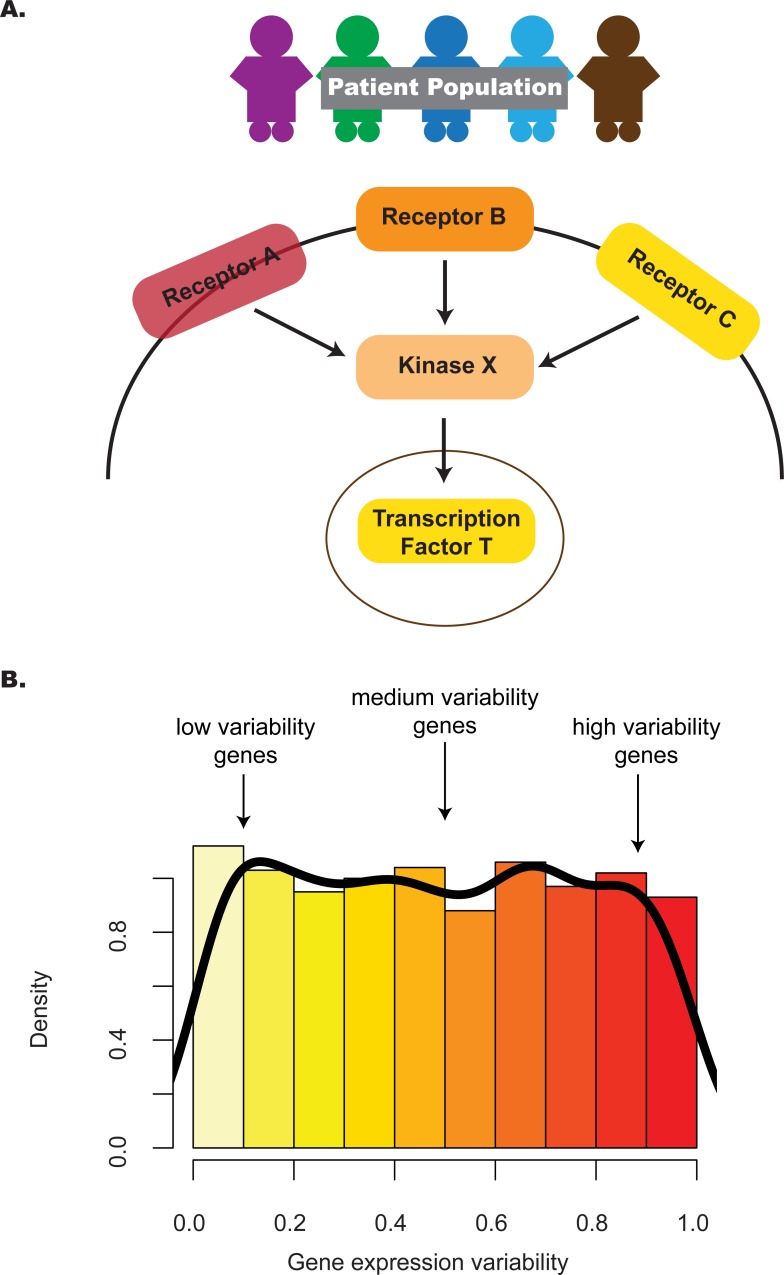
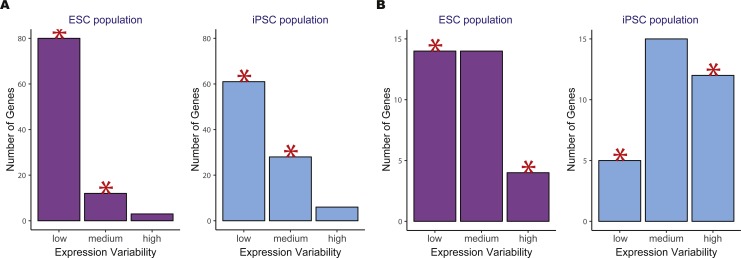
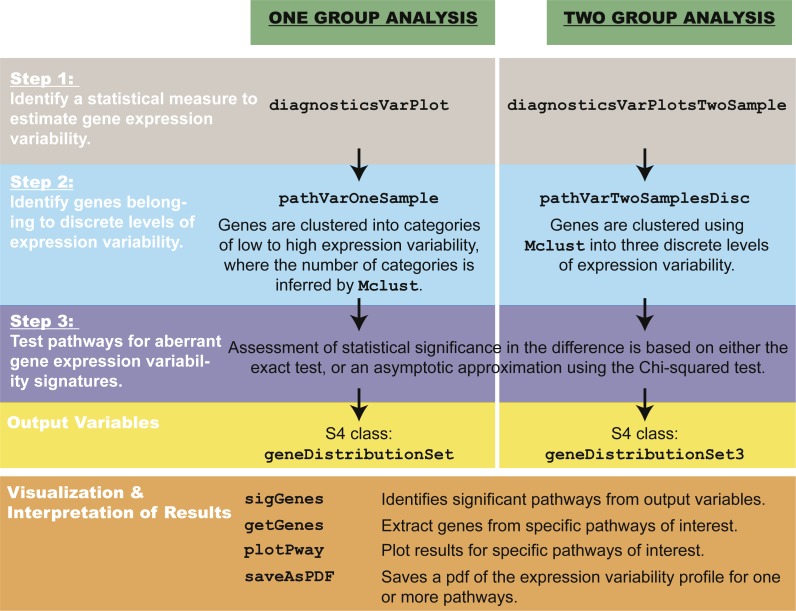
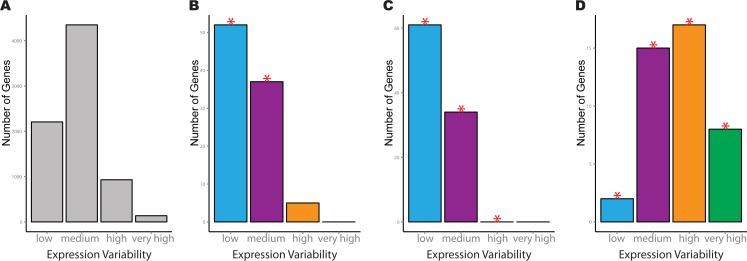
Identifying the pathways that control a cellular phenotype is the first step to building a mechanistic model. Recent examples in developmental biology, cancer genomics, and neurological disease have demonstrated how changes in the variability of gene expression can highlight important genes that are under different degrees of regulatory control. Simple statistical tests exist to identify differentially-variable genes; however, methods for investigating how changes in gene expression variability in the context of pathways and gene sets are under-explored. Here we present a new method that provides functional interpretation of gene expression variability changes at the level of pathways and gene sets. is based on a multinomial exact test, or an asymptotic Chi-squared test as a more computationally-efficient alternative. The method can be used for gene expression studies from any technology platform in all biological settings either with a single phenotypic group, or two-group comparisons. To demonstrate its utility, we applied the method to a diverse set of diseases, species and samples. Results from are benchmarked against analyses based on average expression and two methods of GSEA, and demonstrate that analyses using both statistics are useful for understanding transcriptional regulation. We also provide recommendations for the choice of variability statistic that have been informed through analyses on simulations and real data. Based on the datasets selected, we show how can be used to gain insight into expression variability of single cell versus bulk samples, different stem cell populations, and cancer versus normal tissue comparisons.
识别控制细胞表型的信号通路是构建机制模型的第一步。发育生物学、癌症基因组学和神经疾病领域最近的实例表明,基因表达变异性的变化如何能够突出显示处于不同程度调控之下的重要基因。现有简单的统计测试来识别差异可变基因;然而,在信号通路和基因集背景下研究基因表达变异性变化的方法尚未得到充分探索。在此,我们提出一种新方法,该方法可在信号通路和基因集层面提供基因表达变异性变化的功能解释。该方法基于多项精确检验,或者作为一种计算效率更高的替代方法,基于渐近卡方检验。该方法可用于所有生物学背景下来自任何技术平台的基因表达研究,无论是针对单个表型组,还是两组比较。为证明其效用,我们将该方法应用于多种疾病、物种和样本。所得结果与基于平均表达的分析以及基因集富集分析(GSEA)的两种方法进行了对比,结果表明使用这两种统计方法进行分析均有助于理解转录调控。我们还通过对模拟数据和真实数据的分析,为变异性统计量的选择提供了建议。基于所选数据集,我们展示了该方法如何用于深入了解单细胞与批量样本、不同干细胞群体以及癌症与正常组织比较中的表达变异性。