Department of Pharmacology, The University of Illinois College of Medicine, 835 S. Wolcott Ave. Rm. E403, Chicago, IL, 60612, USA.
Department of Bioengineering, The University of Illinois at Chicago, Chicago, IL, USA.
BMC Bioinformatics. 2018 Jun 7;19(1):217. doi: 10.1186/s12859-018-2190-6.
The heterogeneity of cells across tissue types represents a major challenge for studying biological mechanisms as well as for therapeutic targeting of distinct tissues. Computational prediction of tissue-specific gene regulatory networks may provide important insights into the mechanisms underlying the cellular heterogeneity of cells in distinct organs and tissues.
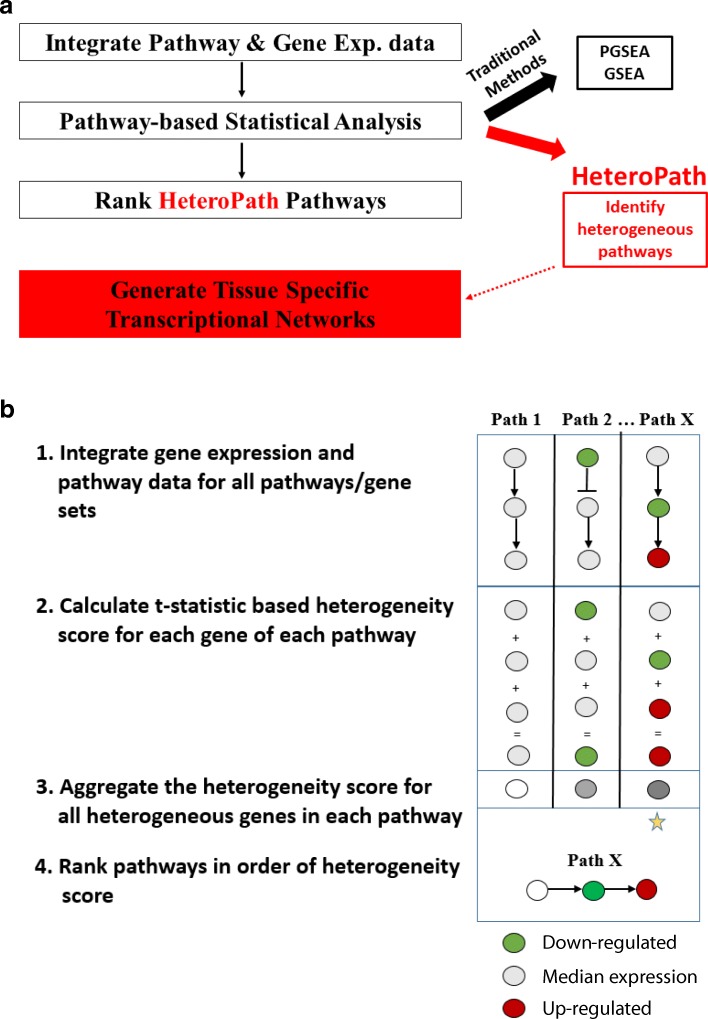
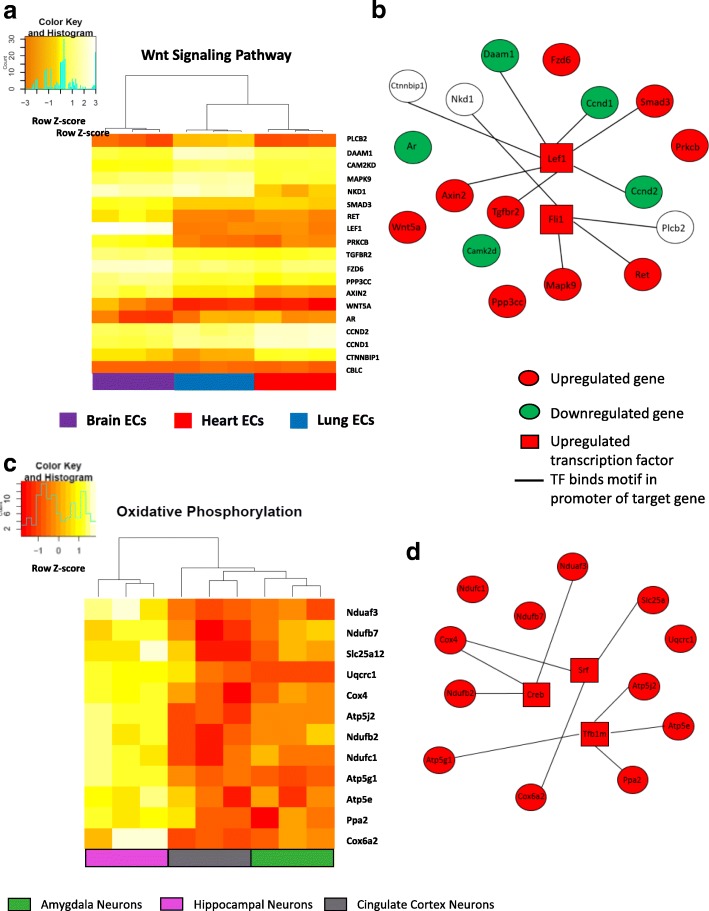
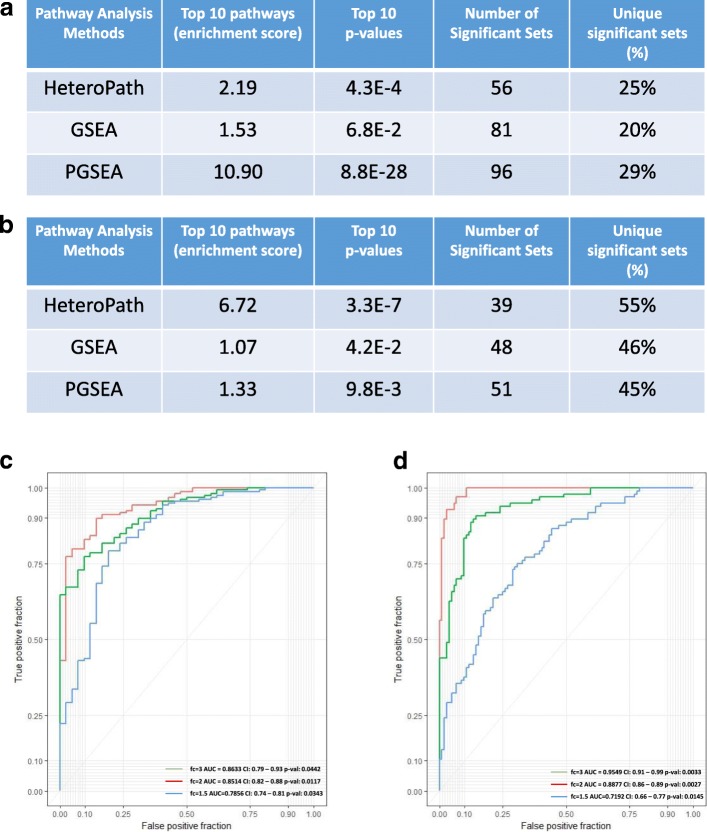
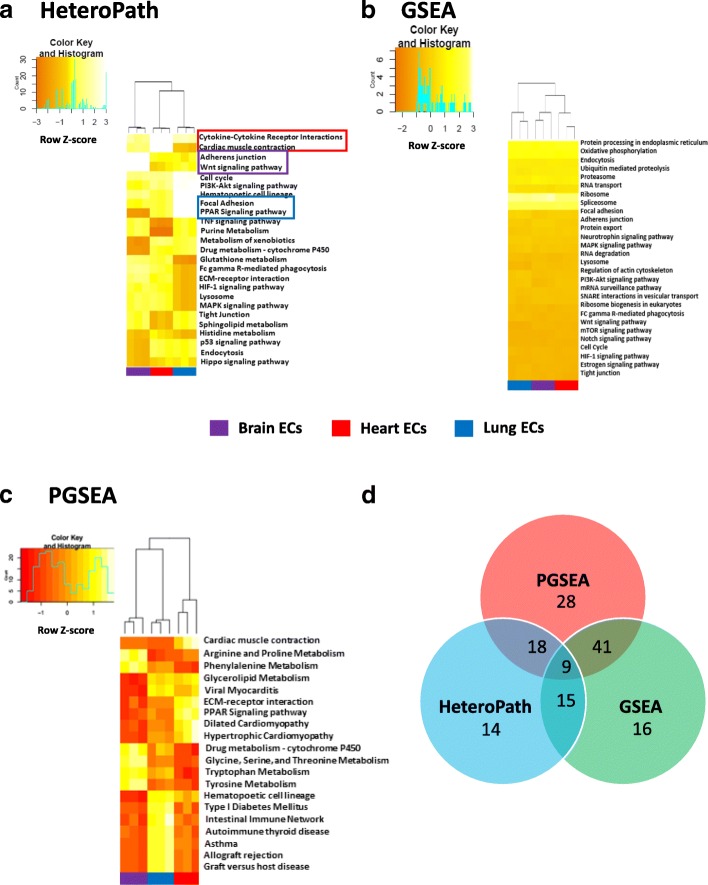
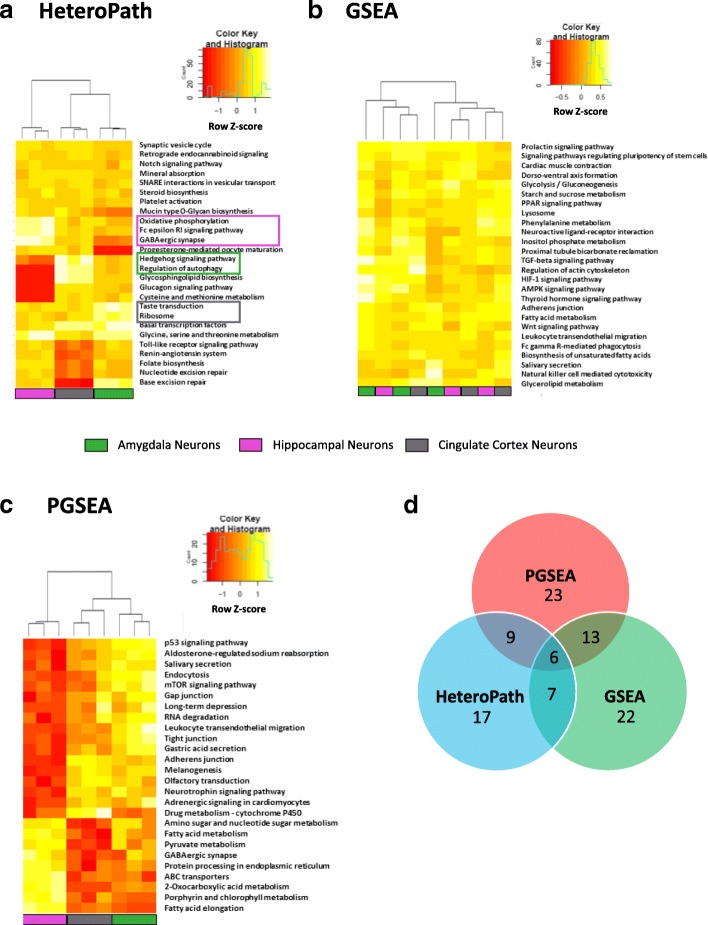
Using three pathway analysis techniques, gene set enrichment analysis (GSEA), parametric analysis of gene set enrichment (PGSEA), alongside our novel model (HeteroPath), which assesses heterogeneously upregulated and downregulated genes within the context of pathways, we generated distinct tissue-specific gene regulatory networks. We analyzed gene expression data derived from freshly isolated heart, brain, and lung endothelial cells and populations of neurons in the hippocampus, cingulate cortex, and amygdala. In both datasets, we found that HeteroPath segregated the distinct cellular populations by identifying regulatory pathways that were not identified by GSEA or PGSEA. Using simulated datasets, HeteroPath demonstrated robustness that was comparable to what was seen using existing gene set enrichment methods. Furthermore, we generated tissue-specific gene regulatory networks involved in vascular heterogeneity and neuronal heterogeneity by performing motif enrichment of the heterogeneous genes identified by HeteroPath and linking the enriched motifs to regulatory transcription factors in the ENCODE database.
HeteroPath assesses contextual bidirectional gene expression within pathways and thus allows for transcriptomic assessment of cellular heterogeneity. Unraveling tissue-specific heterogeneity of gene expression can lead to a better understanding of the molecular underpinnings of tissue-specific phenotypes.
组织类型之间的细胞异质性是研究生物学机制以及针对不同组织进行治疗靶向的主要挑战。计算预测组织特异性基因调控网络可能为深入了解不同器官和组织中细胞的细胞异质性的潜在机制提供重要的见解。
我们使用三种途径分析技术,即基因集富集分析(GSEA)、基因集富集的参数分析(PGSEA)以及我们的新型模型(HeteroPath),评估了在途径背景下异质上调和下调的基因,生成了不同的组织特异性基因调控网络。我们分析了从新鲜分离的心脏、大脑和肺内皮细胞以及海马体、扣带回皮质和杏仁核中的神经元群体中获得的基因表达数据。在这两个数据集上,我们发现 HeteroPath 通过识别 GSEA 或 PGSEA 未识别的调节途径,将不同的细胞群体进行了分类。使用模拟数据集,HeteroPath 表现出了与现有基因集富集方法相当的稳健性。此外,我们通过对 HeteroPath 识别的异质基因进行基序富集,并将富集的基序与 ENCODE 数据库中的调节转录因子联系起来,生成了涉及血管异质性和神经元异质性的组织特异性基因调控网络。
HeteroPath 评估了途径内上下文双向基因表达,从而可以对细胞异质性进行转录组评估。揭示基因表达的组织特异性异质性可以帮助更好地理解组织特异性表型的分子基础。