Max Planck Institute for Evolutionary Biology, August-Thienemann-Str. 2, 24306, Plön, Germany.
Institute for Experimental Medicine, Christian-Albrechts-University of Kiel, Arnold-Heller-Str. 3, 24105, Kiel, Germany.
Microbiome. 2017 Jun 6;5(1):59. doi: 10.1186/s40168-017-0275-5.
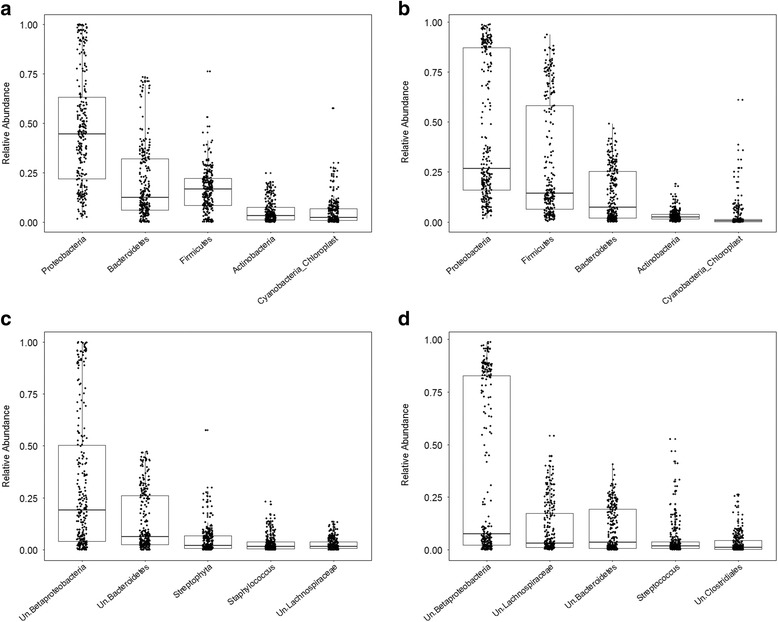
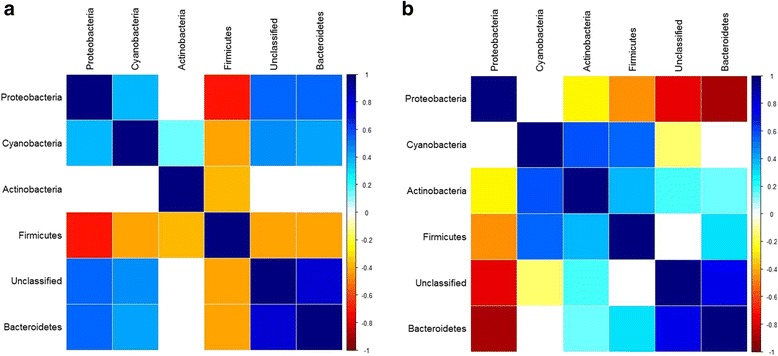
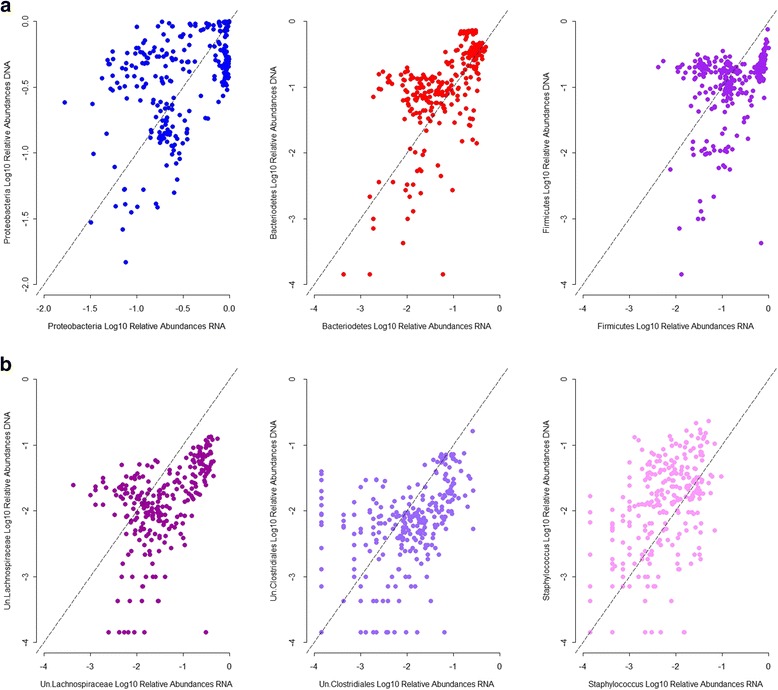
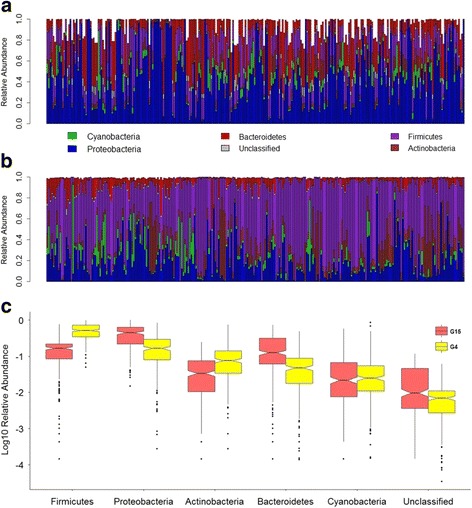
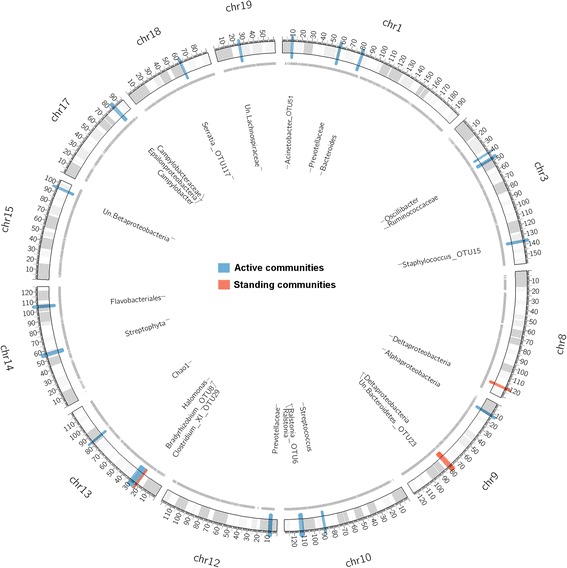
Recent studies highlight the utility of quantitative trait locus (QTL) mapping for determining the contribution of host genetics to interindividual variation in the microbiota. We previously demonstrated that similar to the gut microbiota, abundances of bacterial taxa in the skin are significantly influenced by host genetic variation. In this study, we analyzed the skin microbiota of mice from the 15th generation of an advanced intercross line using a novel approach of extending bacterial trait mapping to both the 16S rRNA gene copy (DNA) and transcript (RNA) levels, which reflect relative bacterial cell number and activity, respectively.
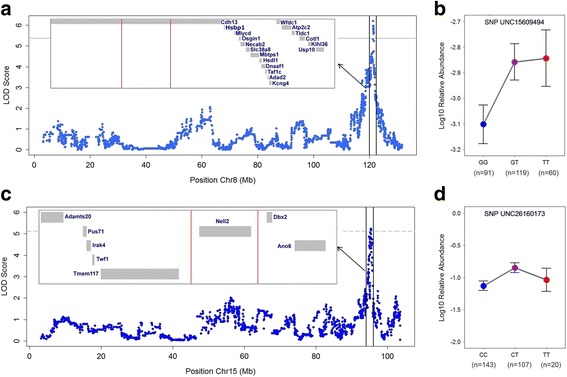
Remarkably, the combination of highly recombined individuals and 53,203 informative SNPs allowed the identification of genomic intervals as small as <0.1 megabases containing single genes. Furthermore, the inclusion of 16S rRNA transcript-level mapping dramatically increased the number of significant associations detected, with five versus 21 significant SNP-bacterial trait associations based on DNA- compared to RNA-level profiling, respectively. Importantly, the genomic intervals identified contain many genes involved in skin inflammation and cancer and are further supported by the bacterial traits they influence, which in some cases have known genotoxic or probiotic capabilities.
These results indicate that profiling based on the relative activity levels of bacterial community members greatly enhances the capability of detecting interactions between the host and its associated microbes. Finally, the identification of several genes involved in skin cancer suggests that similar to colon carcinogenesis, the resident microbiota may play a role in skin cancer susceptibility and its potential prevention and/or treatment.
最近的研究强调了数量性状基因座(QTL)图谱在确定宿主遗传对微生物群落个体间变异的贡献方面的效用。我们之前的研究表明,与肠道微生物群相似,皮肤细菌分类群的丰度也受到宿主遗传变异的显著影响。在这项研究中,我们使用一种新的方法分析了第 15 代高级互交系小鼠的皮肤微生物群,该方法将细菌性状图谱扩展到 16S rRNA 基因拷贝(DNA)和转录本(RNA)水平,分别反映相对细菌细胞数量和活性。
值得注意的是,高度重组个体和 53203 个信息 SNP 的结合允许鉴定出基因组间隔区小至<0.1 兆碱基,其中包含单个基因。此外,包括 16S rRNA 转录水平图谱极大地增加了检测到的显著关联数量,与 DNA 相比,基于 RNA 水平图谱分别检测到 5 个和 21 个显著 SNP-细菌性状关联。重要的是,鉴定出的基因组间隔区包含许多参与皮肤炎症和癌症的基因,并且它们所影响的细菌性状进一步支持了这一点,在某些情况下,这些细菌性状具有已知的遗传毒性或益生菌能力。
这些结果表明,基于细菌群落成员相对活性水平的分析极大地提高了检测宿主与其相关微生物之间相互作用的能力。最后,鉴定出的一些参与皮肤癌的基因表明,与结肠癌发生一样,常驻微生物群可能在皮肤癌易感性及其潜在的预防和/或治疗中发挥作用。