Nagy J, Kobolák J, Berzsenyi S, Ábrahám Z, Avci H X, Bock I, Bekes Z, Hodoscsek B, Chandrasekaran A, Téglási A, Dezső P, Koványi B, Vörös E T, Fodor L, Szél T, Németh K, Balázs A, Dinnyés A, Lendvai B, Lévay G, Román V
Pharmacology and Drug Safety Research, Gedeon Richter Plc., Budapest, Hungary.
BioTalentum Ltd., Gödöllő, Hungary.
Transl Psychiatry. 2017 Jul 25;7(7):e1179. doi: 10.1038/tp.2017.144.
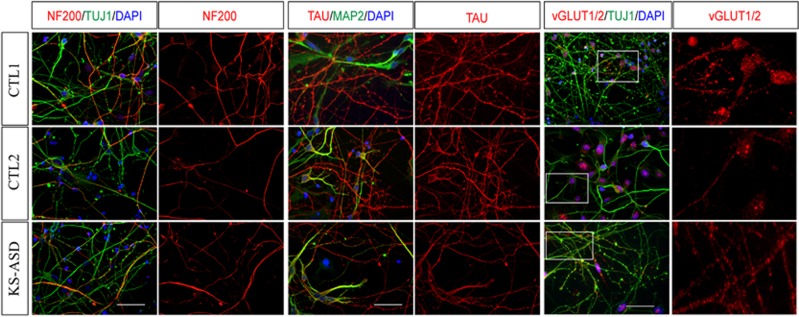
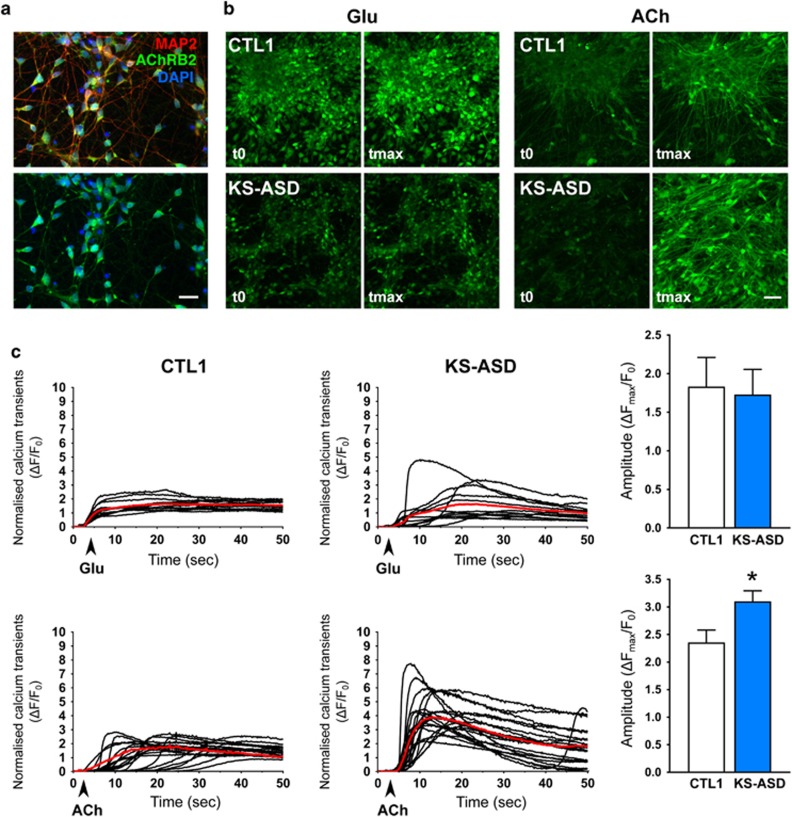
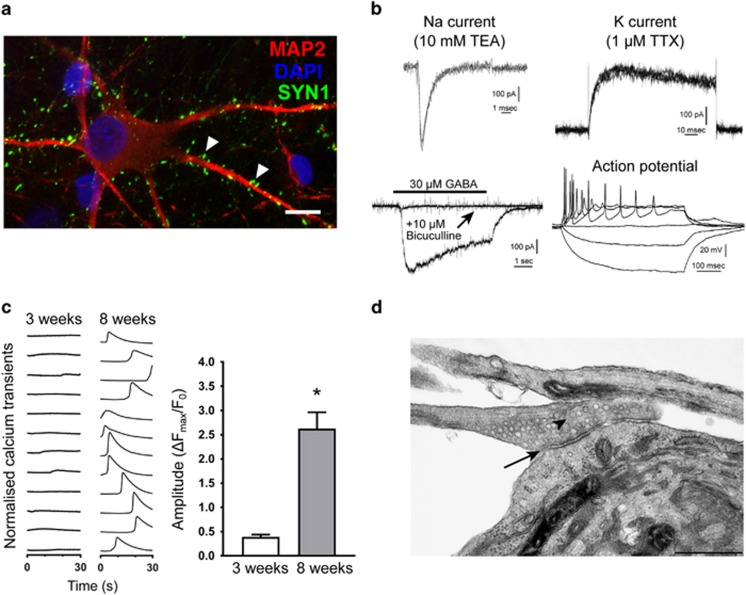
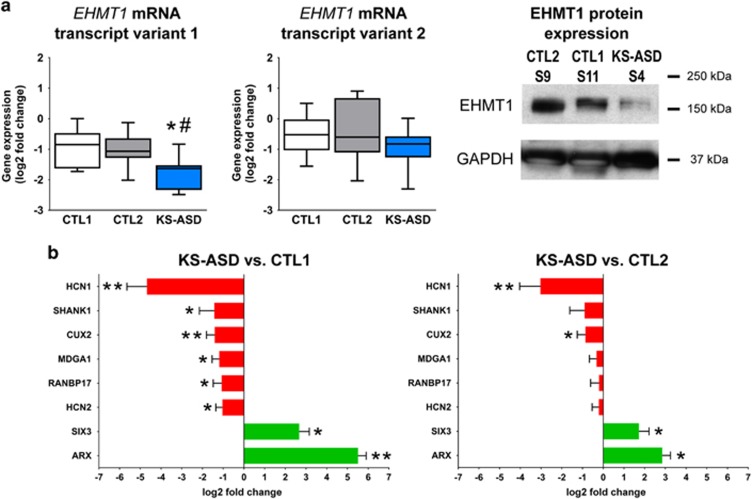
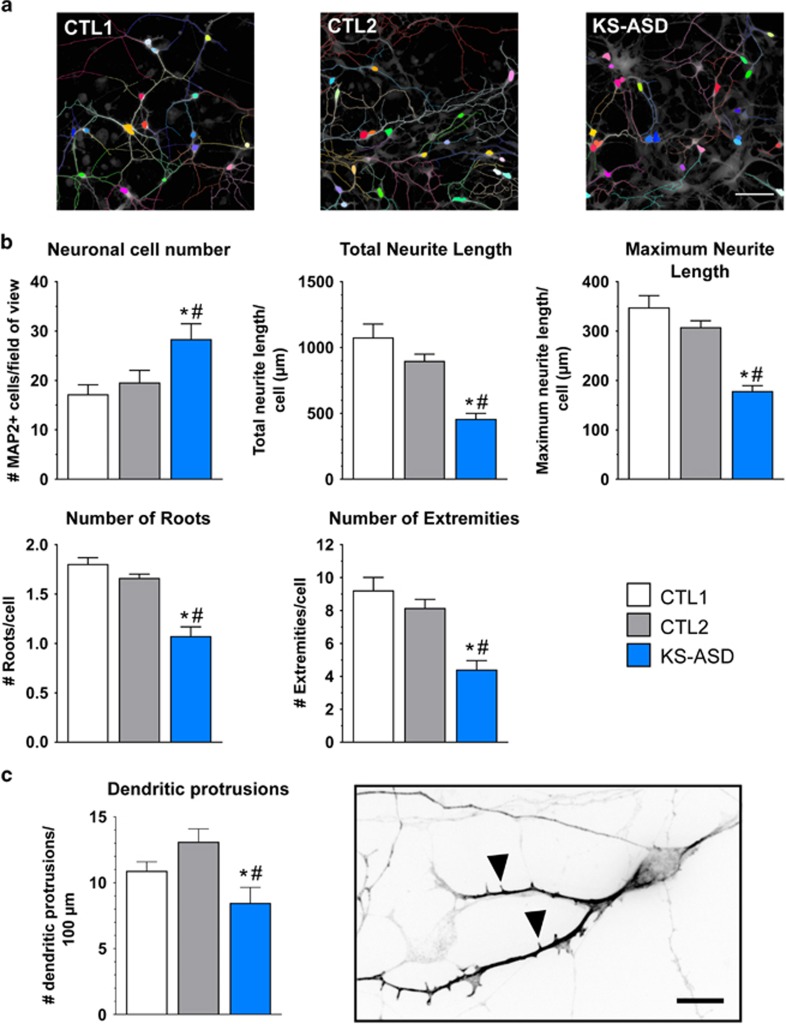
The aim of the present study was to establish an in vitro Kleefstra syndrome (KS) disease model using the human induced pluripotent stem cell (hiPSC) technology. Previously, an autism spectrum disorder (ASD) patient with Kleefstra syndrome (KS-ASD) carrying a deleterious premature termination codon mutation in the EHMT1 gene was identified. Patient specific hiPSCs generated from peripheral blood mononuclear cells of the KS-ASD patient were differentiated into post-mitotic cortical neurons. Lower levels of EHMT1 mRNA as well as protein expression were confirmed in these cells. Morphological analysis on neuronal cells differentiated from the KS-ASD patient-derived hiPSC clones showed significantly shorter neurites and reduced arborization compared to cells generated from healthy controls. Moreover, density of dendritic protrusions of neuronal cells derived from KS-ASD hiPSCs was lower than that of control cells. Synaptic connections and spontaneous neuronal activity measured by live cell calcium imaging could be detected after 5 weeks of differentiation, when KS-ASD cells exhibited higher sensitivity of calcium responses to acetylcholine stimulation indicating a lower nicotinic cholinergic tone at baseline condition in KS-ASD cells. In addition, gene expression profiling of differentiated neuronal cells from the KS-ASD patient revealed higher expression of proliferation-related genes and lower mRNA levels of genes involved in neuronal maturation and migration. Our data demonstrate anomalous neuronal morphology, functional activity and gene expression in KS-ASD patient-specific hiPSC-derived neuronal cultures, which offers an in vitro system that contributes to a better understanding of KS and potentially other neurodevelopmental disorders including ASD.
本研究的目的是利用人类诱导多能干细胞(hiPSC)技术建立一种体外克莱夫斯特拉综合征(KS)疾病模型。此前,已鉴定出一名患有克莱夫斯特拉综合征(KS-ASD)的自闭症谱系障碍(ASD)患者,其EHMT1基因携带有害的过早终止密码子突变。从KS-ASD患者的外周血单核细胞中产生的患者特异性hiPSC被分化为有丝分裂后皮质神经元。在这些细胞中证实了EHMT1 mRNA水平以及蛋白质表达较低。对从KS-ASD患者来源的hiPSC克隆分化而来的神经元细胞进行的形态学分析显示,与从健康对照中产生的细胞相比,神经突明显更短且分支减少。此外,来自KS-ASD hiPSC的神经元细胞的树突突起密度低于对照细胞。分化5周后,可以检测到通过活细胞钙成像测量的突触连接和自发神经元活动,此时KS-ASD细胞对乙酰胆碱刺激表现出更高的钙反应敏感性,表明KS-ASD细胞在基线条件下烟碱胆碱能张力较低。此外,对来自KS-ASD患者的分化神经元细胞进行的基因表达谱分析显示,增殖相关基因表达较高,而参与神经元成熟和迁移的基因的mRNA水平较低。我们的数据证明了KS-ASD患者特异性hiPSC来源的神经元培养物中存在异常的神经元形态、功能活动和基因表达,这提供了一个体外系统,有助于更好地理解KS以及潜在的其他神经发育障碍,包括ASD。