Ma Shisong, Ding Zehong, Li Pinghua
School of Life Sciences, University of Science and Technology of China, Hefei, Anhui, China.
The Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences, Haikou, Hainan, China.
BMC Plant Biol. 2017 Aug 1;17(1):131. doi: 10.1186/s12870-017-1077-4.
The advent of big data in biology offers opportunities while poses challenges to derive biological insights. For maize, a large amount of publicly available transcriptome datasets have been generated but a comprehensive analysis is lacking.
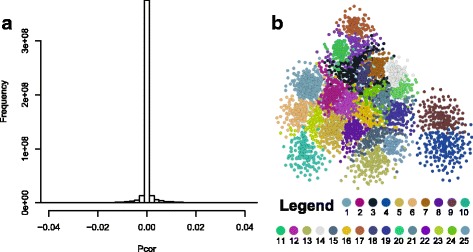
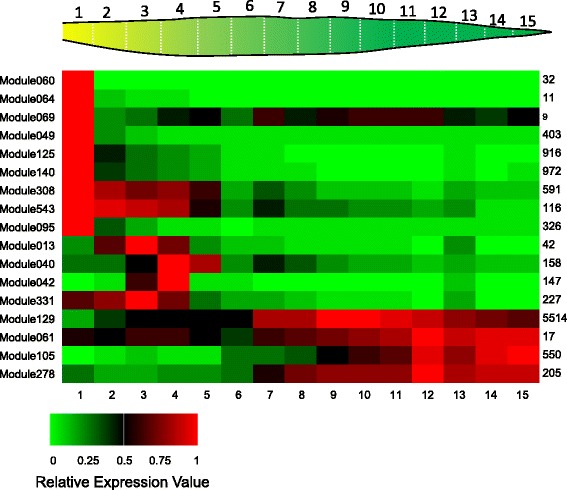
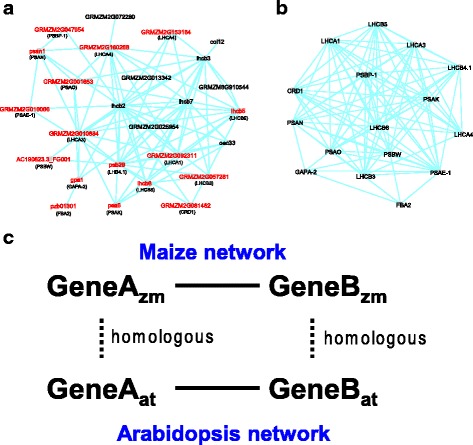
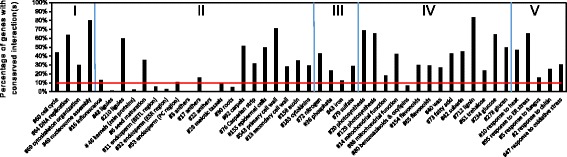
We constructed a maize gene co-expression network based on the graphical Gaussian model, using massive RNA-seq data. The network, containing 20,269 genes, assembles into 964 gene modules that function in a variety of plant processes, such as cell organization, the development of inflorescences, ligules and kernels, the uptake and utilization of nutrients (e.g. nitrogen and phosphate), the metabolism of benzoxazionids, oxylipins, flavonoids, and wax, and the response to stresses. Among them, the inflorescences development module is enriched with domestication genes (like ra1, ba1, gt1, tb1, tga1) that control plant architecture and kernel structure, while multiple other modules relate to diverse agronomic traits. Contained within these modules are transcription factors acting as known or potential expression regulators for the genes within the same modules, suggesting them as candidate regulators for related biological processes. A comparison with an established Arabidopsis network revealed conserved gene association patterns for specific modules involved in cell organization, nutrients uptake & utilization, and metabolism. The analysis also identified significant divergences between the two species for modules that orchestrate developmental pathways.
This network sheds light on how gene modules are organized between different species in the context of evolutionary divergence and highlights modules whose structure and gene content can provide important resources for maize gene functional studies with application potential.
生物学中大数据的出现带来了机遇,但也对获取生物学见解提出了挑战。对于玉米而言,已产生了大量可公开获取的转录组数据集,但缺乏全面分析。
我们基于图形高斯模型,利用大量RNA测序数据构建了一个玉米基因共表达网络。该网络包含20269个基因,组装成964个基因模块,这些模块在多种植物过程中发挥作用,如细胞组织、花序、叶舌和籽粒的发育、养分(如氮和磷)的吸收和利用、苯并恶唑嗪酮、氧脂、类黄酮和蜡的代谢以及对胁迫的响应。其中,花序发育模块富含控制植物结构和籽粒结构的驯化基因(如ra1、ba1、gt1、tb1、tga1),而其他多个模块与多种农艺性状相关。这些模块中包含转录因子,它们作为同一模块内基因的已知或潜在表达调节因子,表明它们是相关生物学过程的候选调节因子。与已建立的拟南芥网络进行比较,揭示了在细胞组织、养分吸收与利用以及代谢等特定模块中保守的基因关联模式。该分析还确定了在协调发育途径的模块上两个物种之间的显著差异。
该网络揭示了在进化分歧背景下不同物种间基因模块是如何组织的,并突出了其结构和基因内容可为具有应用潜力的玉米基因功能研究提供重要资源的模块。