Langmead Ben
Department of Computer Science, Whiting School of Engineering, Johns Hopkins University, 3400 North Charles St, Baltimore, 21218-2682, USA.
Department of Biostatistics, Bloomberg School of Public Health, Johns Hopkins University, 615 N Wolfe St, Baltimore, 21205, USA.
Genome Biol. 2017 Aug 10;18(1):152. doi: 10.1186/s13059-017-1290-3.
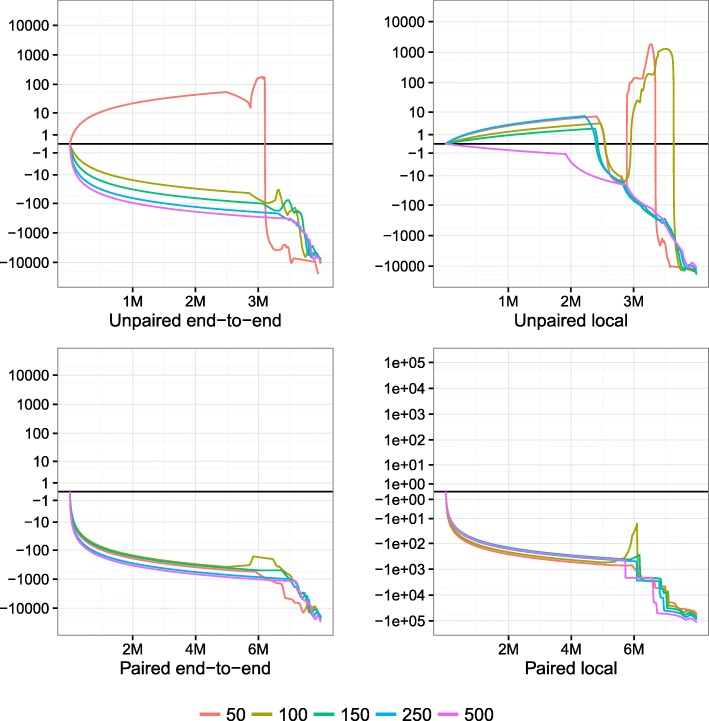
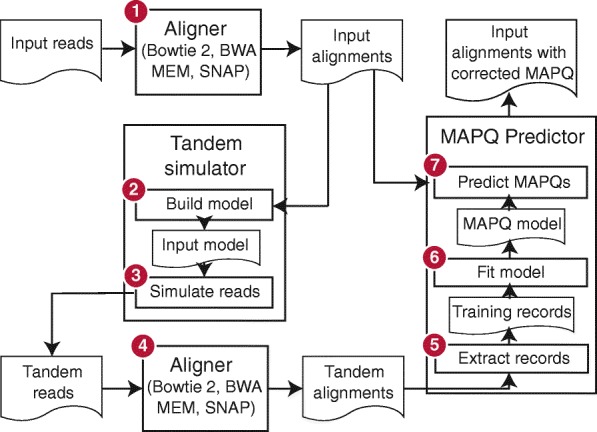
Read alignment is the first step in most sequencing data analyses. Because a read's point of origin can be ambiguous, aligners report a mapping quality, which is the probability that the reported alignment is incorrect. Despite its importance, there is no established and general method for calculating mapping quality. I describe a framework for predicting mapping qualities that works by simulating a set of tandem reads. These are like the input reads in important ways, but the true point of origin is known. I implement this method in an accurate and low-overhead tool called Qtip, which is compatible with popular aligners.
读取比对是大多数测序数据分析的第一步。由于读取的起源点可能不明确,比对工具会报告一个映射质量,即所报告的比对不正确的概率。尽管其很重要,但目前尚无既定的通用方法来计算映射质量。我描述了一个通过模拟一组串联读取来预测映射质量的框架。这些串联读取在重要方面与输入读取相似,但真实的起源点是已知的。我在一个名为Qtip的准确且低开销的工具中实现了此方法,该工具与流行的比对工具兼容。