Interuniversity Institute of Bioinformatics in Brussels, ULB/VUB, Triomflaan, BC building, 6th floor, CP 263, 1050, Brussels, Belgium.
Machine Learning Group, Université Libre de Bruxelles, Boulevard du Triomphe, CP 212, 1050, Brussels, Belgium.
Sci Rep. 2017 Aug 18;7(1):8826. doi: 10.1038/s41598-017-08366-3.
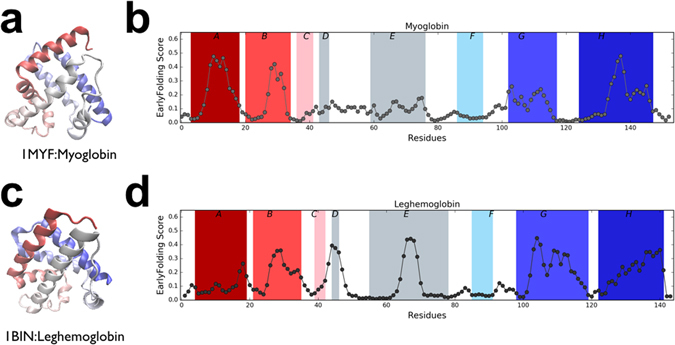
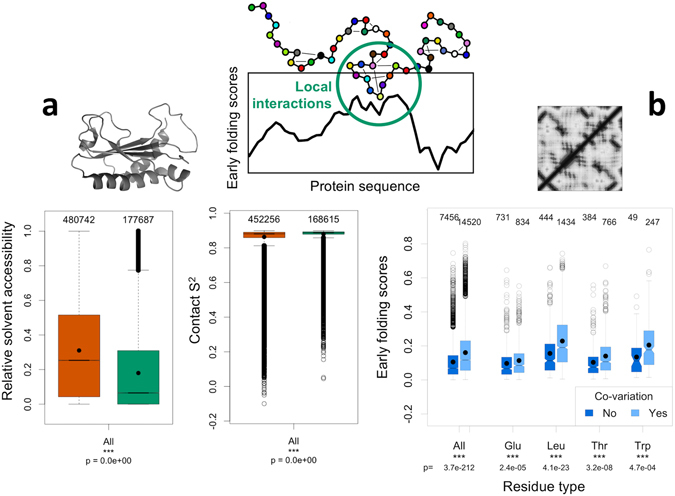
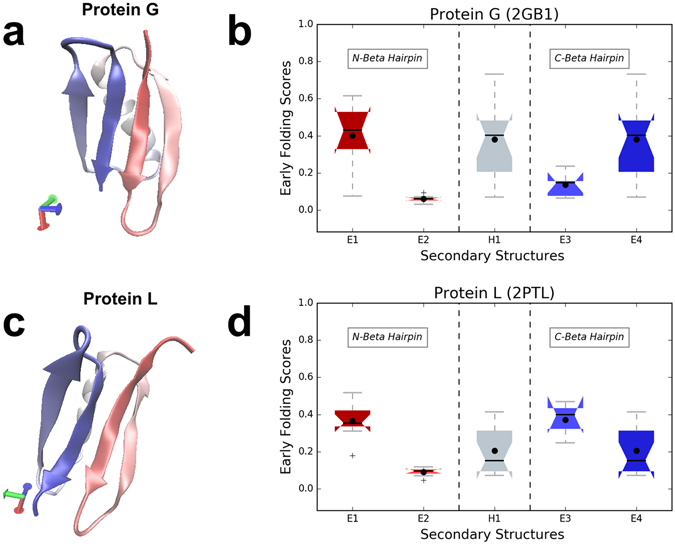
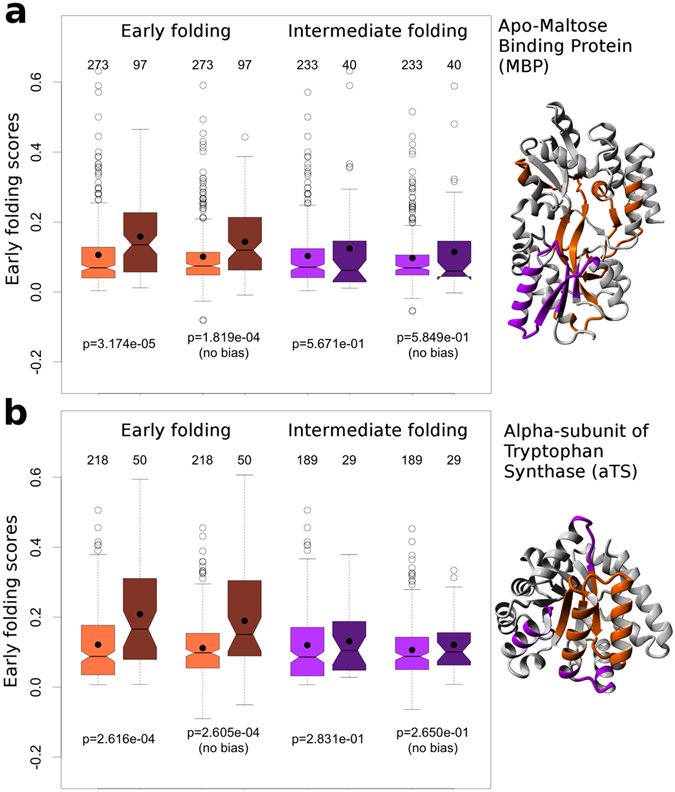
Protein folding is a complex process that can lead to disease when it fails. Especially poorly understood are the very early stages of protein folding, which are likely defined by intrinsic local interactions between amino acids close to each other in the protein sequence. We here present EFoldMine, a method that predicts, from the primary amino acid sequence of a protein, which amino acids are likely involved in early folding events. The method is based on early folding data from hydrogen deuterium exchange (HDX) data from NMR pulsed labelling experiments, and uses backbone and sidechain dynamics as well as secondary structure propensities as features. The EFoldMine predictions give insights into the folding process, as illustrated by a qualitative comparison with independent experimental observations. Furthermore, on a quantitative proteome scale, the predicted early folding residues tend to become the residues that interact the most in the folded structure, and they are often residues that display evolutionary covariation. The connection of the EFoldMine predictions with both folding pathway data and the folded protein structure suggests that the initial statistical behavior of the protein chain with respect to local structure formation has a lasting effect on its subsequent states.
蛋白质折叠是一个复杂的过程,当它失败时可能会导致疾病。特别是蛋白质折叠的早期阶段,其非常难以理解,这些阶段可能由蛋白质序列中彼此靠近的氨基酸之间的固有局部相互作用来定义。我们在这里介绍 EFoldMine,这是一种从蛋白质的一级氨基酸序列预测哪些氨基酸可能参与早期折叠事件的方法。该方法基于来自 NMR 脉冲标记实验的氢氘交换 (HDX) 数据的早期折叠数据,并将骨架和侧链动力学以及二级结构倾向作为特征。EFoldMine 的预测提供了对折叠过程的深入了解,这可以通过与独立实验观察的定性比较来说明。此外,在定量蛋白质组范围内,预测的早期折叠残基往往成为在折叠结构中相互作用最多的残基,并且它们通常是显示进化共变的残基。EFoldMine 预测与折叠途径数据和折叠蛋白质结构的连接表明,蛋白质链相对于局部结构形成的初始统计行为对其后续状态具有持久影响。