Ameur Adam, Dahlberg Johan, Olason Pall, Vezzi Francesco, Karlsson Robert, Martin Marcel, Viklund Johan, Kähäri Andreas Kusalananda, Lundin Pär, Che Huiwen, Thutkawkorapin Jessada, Eisfeldt Jesper, Lampa Samuel, Dahlberg Mats, Hagberg Jonas, Jareborg Niclas, Liljedahl Ulrika, Jonasson Inger, Johansson Åsa, Feuk Lars, Lundeberg Joakim, Syvänen Ann-Christine, Lundin Sverker, Nilsson Daniel, Nystedt Björn, Magnusson Patrik Ke, Gyllensten Ulf
Science for Life Laboratory, Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden.
National Genomics Infrastructure, Science for Life Laboratory, Sweden.
Eur J Hum Genet. 2017 Nov;25(11):1253-1260. doi: 10.1038/ejhg.2017.130. Epub 2017 Aug 23.
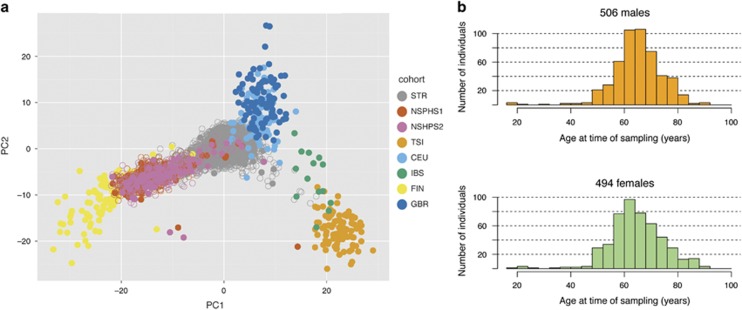
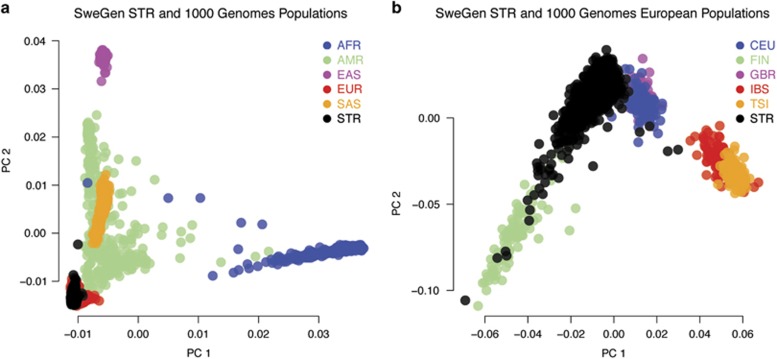
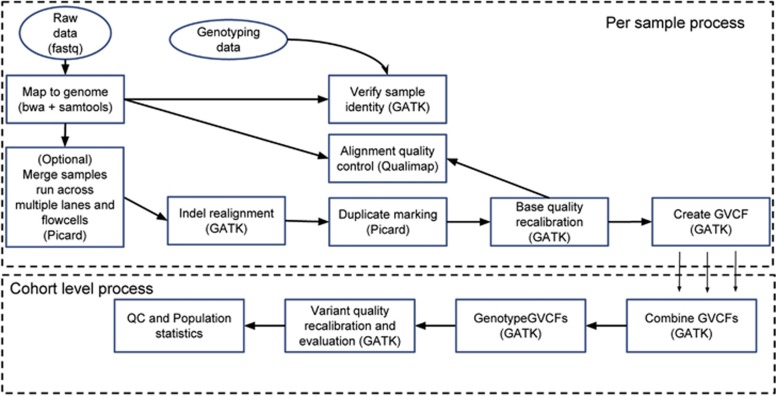
Here we describe the SweGen data set, a comprehensive map of genetic variation in the Swedish population. These data represent a basic resource for clinical genetics laboratories as well as for sequencing-based association studies by providing information on genetic variant frequencies in a cohort that is well matched to national patient cohorts. To select samples for this study, we first examined the genetic structure of the Swedish population using high-density SNP-array data from a nation-wide cohort of over 10 000 Swedish-born individuals included in the Swedish Twin Registry. A total of 1000 individuals, reflecting a cross-section of the population and capturing the main genetic structure, were selected for whole-genome sequencing. Analysis pipelines were developed for automated alignment, variant calling and quality control of the sequencing data. This resulted in a genome-wide collection of aggregated variant frequencies in the Swedish population that we have made available to the scientific community through the website https://swefreq.nbis.se. A total of 29.2 million single-nucleotide variants and 3.8 million indels were detected in the 1000 samples, with 9.9 million of these variants not present in current databases. Each sample contributed with an average of 7199 individual-specific variants. In addition, an average of 8645 larger structural variants (SVs) were detected per individual, and we demonstrate that the population frequencies of these SVs can be used for efficient filtering analyses. Finally, our results show that the genetic diversity within Sweden is substantial compared with the diversity among continental European populations, underscoring the relevance of establishing a local reference data set.
在此,我们描述了SweGen数据集,这是瑞典人群遗传变异的综合图谱。这些数据通过提供与全国患者队列高度匹配的队列中遗传变异频率的信息,为临床遗传学实验室以及基于测序的关联研究提供了基础资源。为了选择本研究的样本,我们首先使用来自瑞典双胞胎登记处的全国范围内超过10000名瑞典出生个体的高密度SNP阵列数据,研究了瑞典人群的遗传结构。总共选择了1000名个体进行全基因组测序,这些个体反映了人群的横断面并捕捉了主要的遗传结构。我们开发了分析流程,用于对测序数据进行自动比对、变异检测和质量控制。这产生了瑞典人群中全基因组汇总变异频率的集合,我们已通过网站https://swefreq.nbis.se向科学界公开。在1000个样本中总共检测到2920万个单核苷酸变异和380万个插入缺失,其中990万个变异在当前数据库中不存在。每个样本平均贡献7199个个体特异性变异。此外,每个个体平均检测到8645个更大的结构变异(SVs),并且我们证明这些SVs的人群频率可用于高效的过滤分析。最后,我们的结果表明,与欧洲大陆人群之间的多样性相比,瑞典国内的遗传多样性相当大,这突出了建立本地参考数据集的重要性。