Department of Computer Science and Informatics, Indiana University, Bloomington, IN, USA.
Department of Psychiatry, University of California San Diego, La Jolla, CA, USA.
Bioinformatics. 2017 Jul 15;33(14):i389-i398. doi: 10.1093/bioinformatics/btx272.
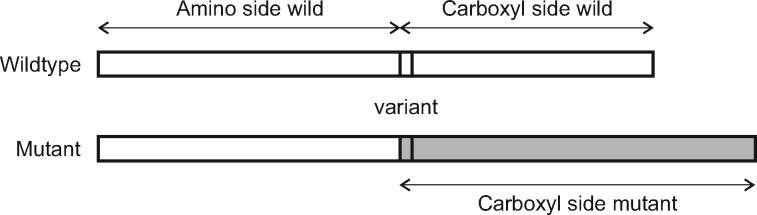
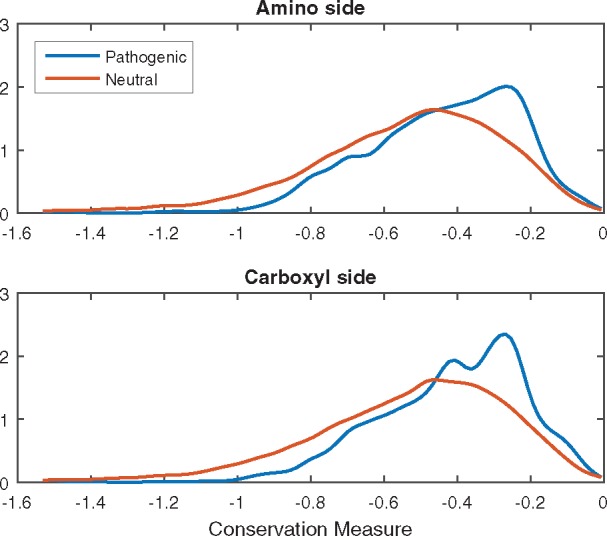
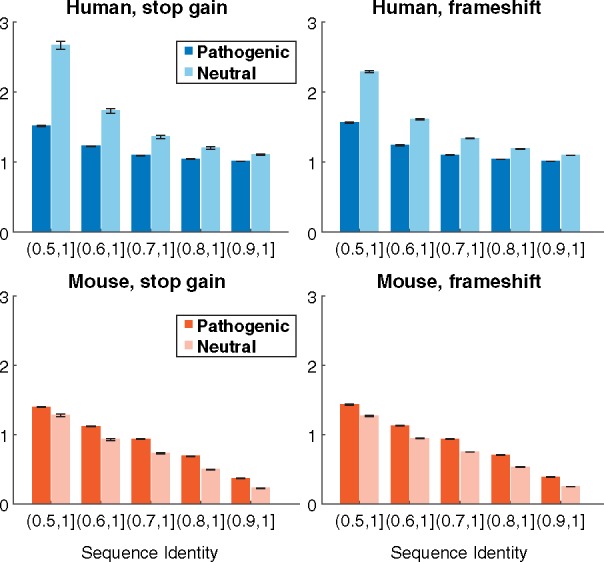
Loss-of-function genetic variants are frequently associated with severe clinical phenotypes, yet many are present in the genomes of healthy individuals. The available methods to assess the impact of these variants rely primarily upon evolutionary conservation with little to no consideration of the structural and functional implications for the protein. They further do not provide information to the user regarding specific molecular alterations potentially causative of disease.
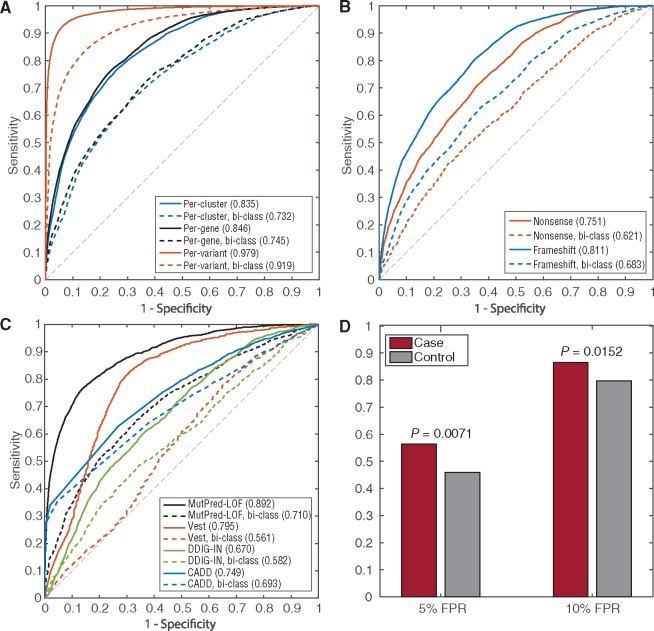
To address this, we investigate protein features underlying loss-of-function genetic variation and develop a machine learning method, MutPred-LOF, for the discrimination of pathogenic and tolerated variants that can also generate hypotheses on specific molecular events disrupted by the variant. We investigate a large set of human variants derived from the Human Gene Mutation Database, ClinVar and the Exome Aggregation Consortium. Our prediction method shows an area under the Receiver Operating Characteristic curve of 0.85 for all loss-of-function variants and 0.75 for proteins in which both pathogenic and neutral variants have been observed. We applied MutPred-LOF to a set of 1142 de novo vari3ants from neurodevelopmental disorders and find enrichment of pathogenic variants in affected individuals. Overall, our results highlight the potential of computational tools to elucidate causal mechanisms underlying loss of protein function in loss-of-function variants.
失能性遗传变异经常与严重的临床表型相关联,但许多变异存在于健康个体的基因组中。现有的评估这些变异影响的方法主要依赖于进化保守性,很少考虑到蛋白质的结构和功能影响。它们也没有为用户提供有关可能导致疾病的特定分子改变的信息。
为了解决这个问题,我们研究了失能性遗传变异所涉及的蛋白质特征,并开发了一种机器学习方法 MutPred-LOF,用于区分致病性和耐受性变异,还可以生成关于变异所破坏的特定分子事件的假设。我们研究了一组来自人类基因突变数据库、ClinVar 和外显子聚集联盟的人类变体。我们的预测方法在所有失能性变异中获得了 0.85 的接收器操作特征曲线下面积,在观察到致病性和中性变异的蛋白质中获得了 0.75 的面积。我们将 MutPred-LOF 应用于一组来自神经发育障碍的 1142 个新发变异体,发现致病性变异体在受影响个体中富集。总体而言,我们的结果强调了计算工具在阐明失能性变异中蛋白质功能丧失的潜在因果机制方面的潜力。