Institute for Molecular Modeling and Simulation, Department for Material Sciences and Process Engineering, University of Natural Resources and Life Sciences (BOKU) Vienna , Muthgasse 18, 1190 Vienna, Austria.
J Chem Theory Comput. 2017 Nov 14;13(11):5697-5708. doi: 10.1021/acs.jctc.7b00706. Epub 2017 Oct 3.
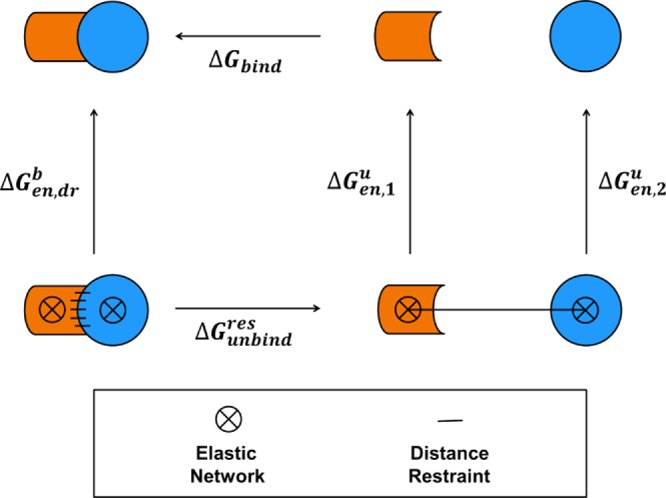
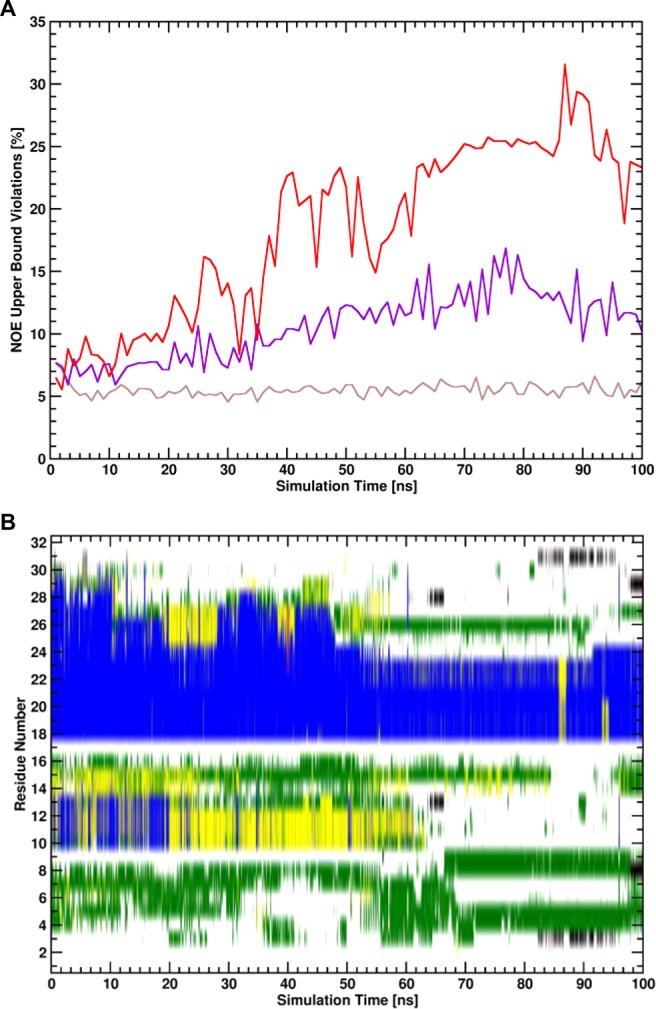
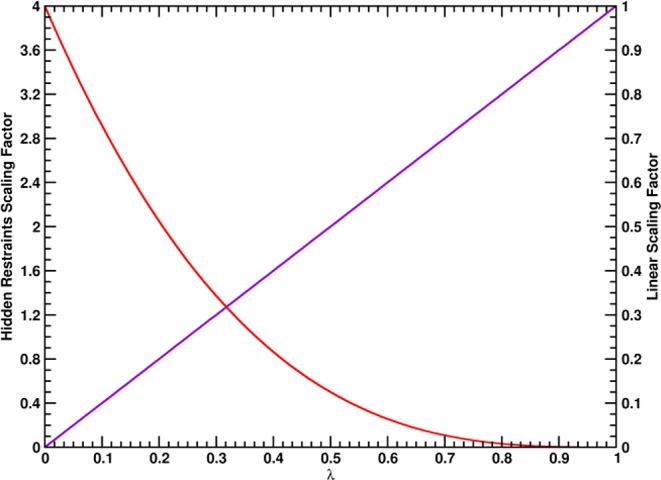
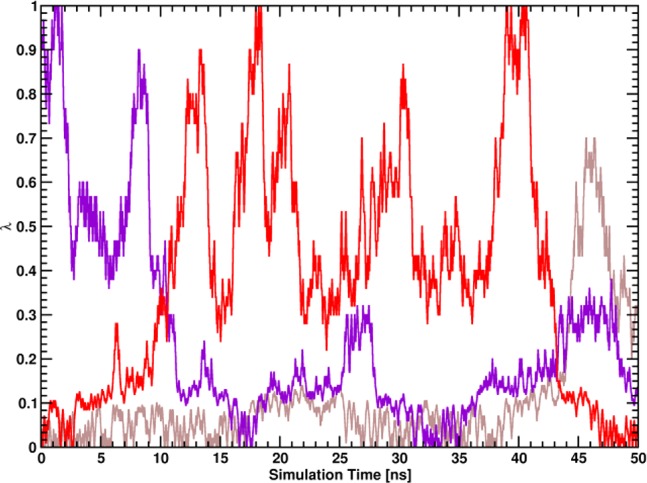
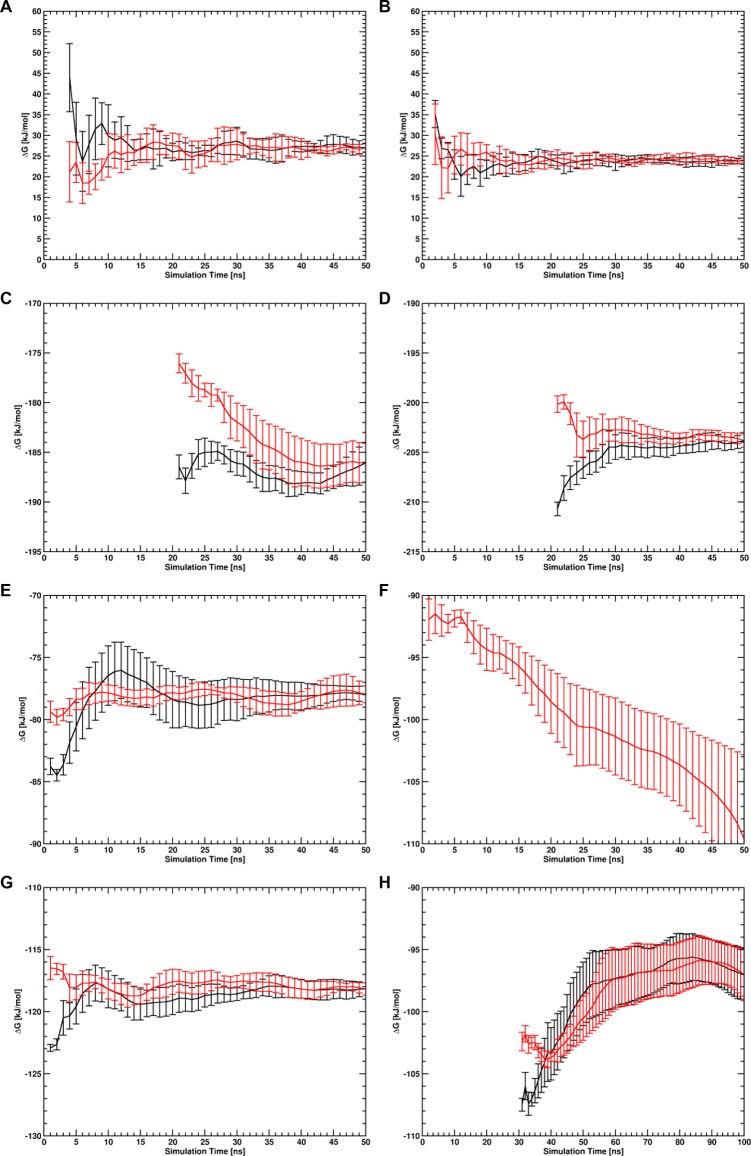
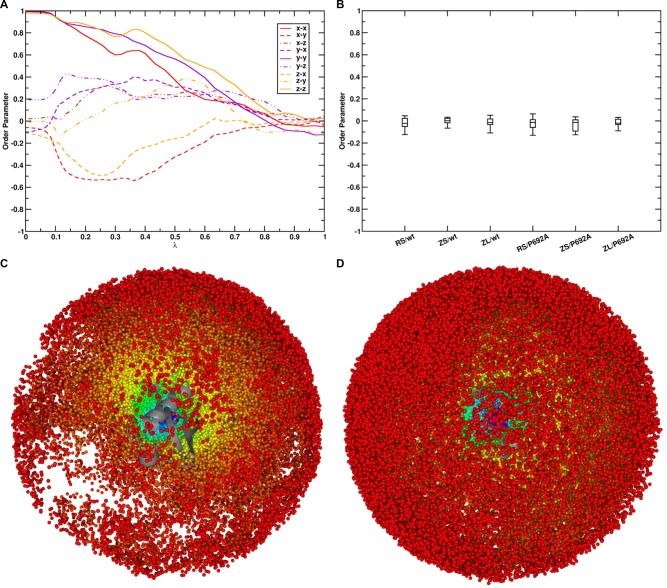
Virtually all biological processes depend on the interaction between proteins at some point. The correct prediction of biomolecular binding free-energies has many interesting applications in both basic and applied pharmaceutical research. While recent advances in the field of molecular dynamics (MD) simulations have proven the feasibility of the calculation of protein-protein binding free energies, the large conformational freedom of proteins and complex free energy landscapes of binding processes make such calculations a difficult task. Moreover, convergence and reversibility of resulting free-energy values remain poorly described. In this work, an easy-to-use, yet robust approach for the calculation of standard-state protein-protein binding free energies using perturbed distance restraints is described. In the binding process the conformations of the proteins were restrained, as suggested earlier. Two approaches to avoid end-state problems upon release of the conformational restraints were compared. The method was evaluated by practical application to a small model complex of ubiquitin and the very flexible ubiquitin-binding domain of human DNA polymerase ι (UBM2). All computed free energy differences were closely monitored for convergence, and the calculated binding free energies had a mean unsigned deviation of only 1.4 or 2.5 kJ·mol from experimental values. Statistical error estimates were in the order of thermal noise. We conclude that the presented method has promising potential for broad applicability to quantitatively describe protein-protein and various other kinds of complex formation.
几乎所有的生物过程都依赖于蛋白质在某个点的相互作用。正确预测生物分子结合自由能在基础和应用药物研究中都有许多有趣的应用。虽然分子动力学 (MD) 模拟领域的最新进展已经证明了计算蛋白质-蛋白质结合自由能的可行性,但蛋白质的构象自由度大,结合过程的自由能景观复杂,使得此类计算成为一项艰巨的任务。此外,所得自由能值的收敛性和可逆性仍描述得很差。在这项工作中,描述了一种使用扰动距离约束计算标准态蛋白质-蛋白质结合自由能的简单但稳健的方法。在结合过程中,如前所述,限制蛋白质的构象。比较了两种避免构象约束释放时出现末端状态问题的方法。该方法通过实际应用于小模型复合物泛素和人类 DNA 聚合酶 ι 的非常灵活的泛素结合域 (UBM2) 进行了评估。所有计算的自由能差异都密切监测收敛性,并且计算的结合自由能与实验值的平均无符号偏差仅为 1.4 或 2.5 kJ·mol。统计误差估计处于热噪声的顺序。我们得出的结论是,所提出的方法具有广泛应用于定量描述蛋白质-蛋白质和各种其他类型的复杂形成的巨大潜力。