Li Xiao, Zhou Bing, Chen Liang, Gou Lan-Tao, Li Hairi, Fu Xiang-Dong
Department of Cellular and Molecular Medicine, University of California, San Diego, La Jolla, California, USA.
Institute of Genomic Medicine, University of California, San Diego, La Jolla, California, USA.
Nat Biotechnol. 2017 Oct;35(10):940-950. doi: 10.1038/nbt.3968. Epub 2017 Sep 18.
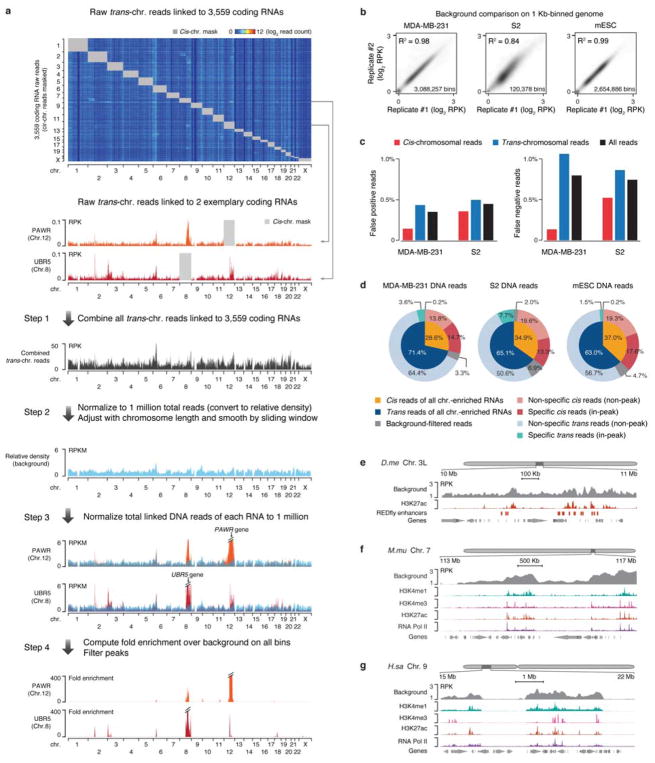
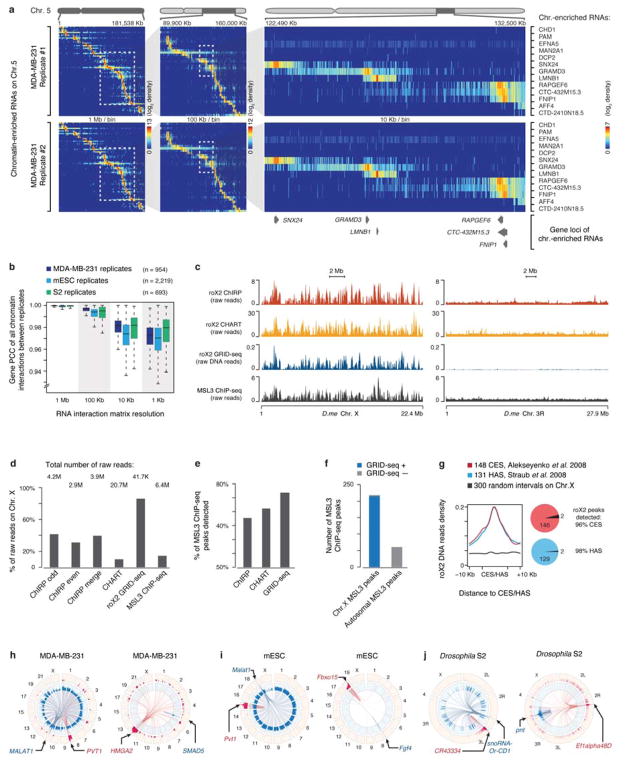
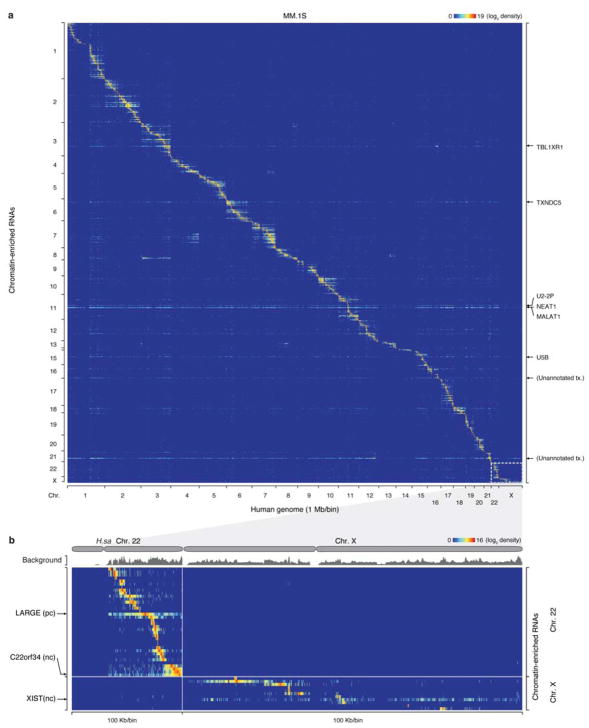
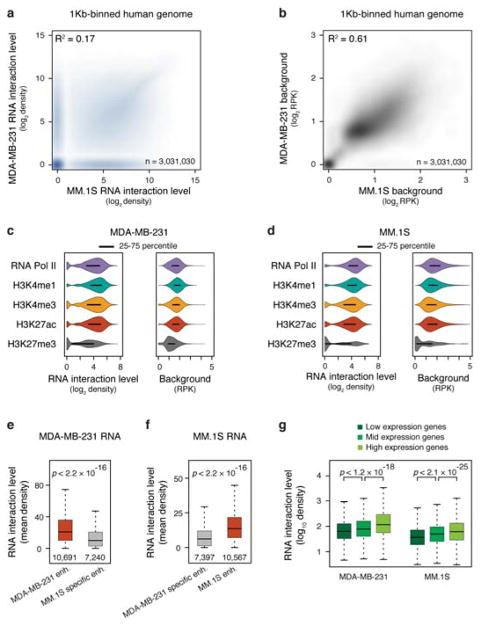
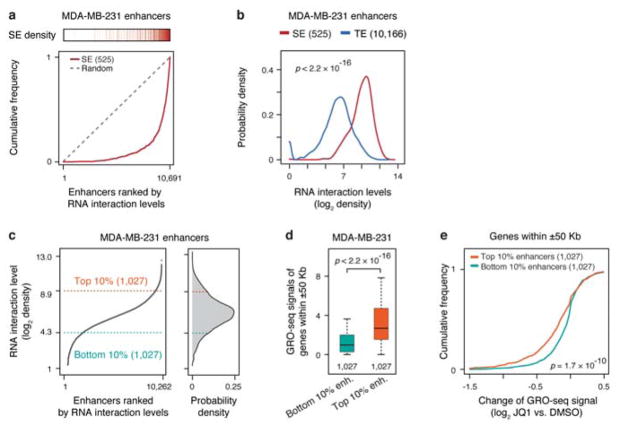
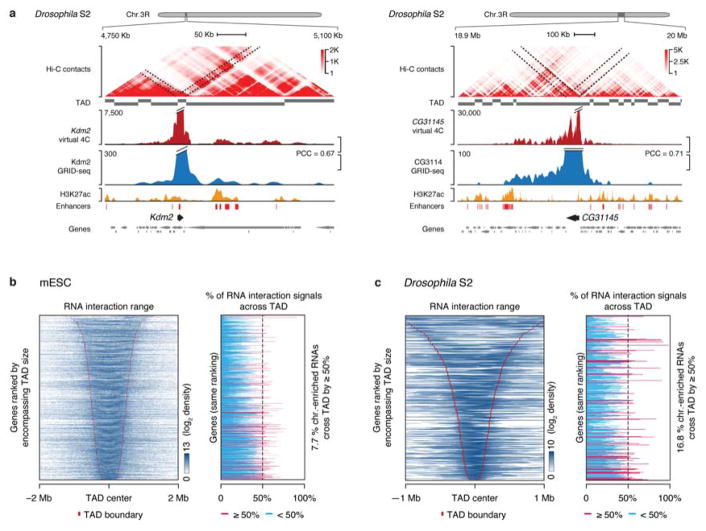
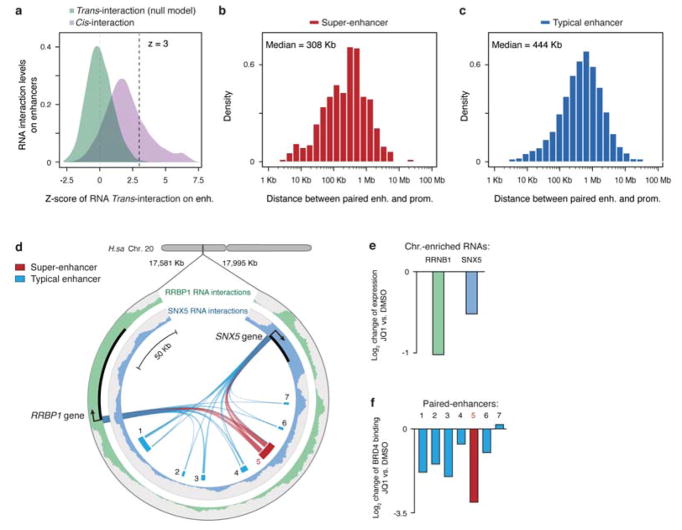
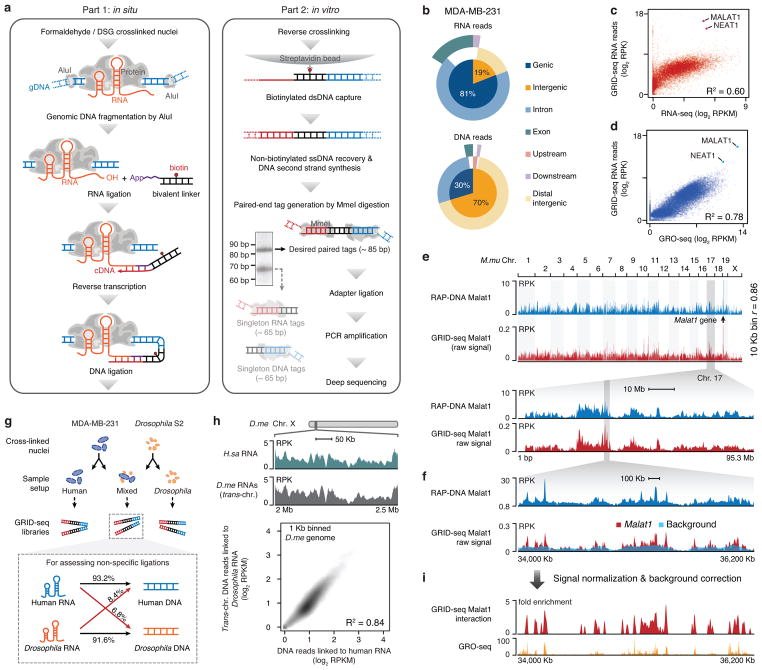
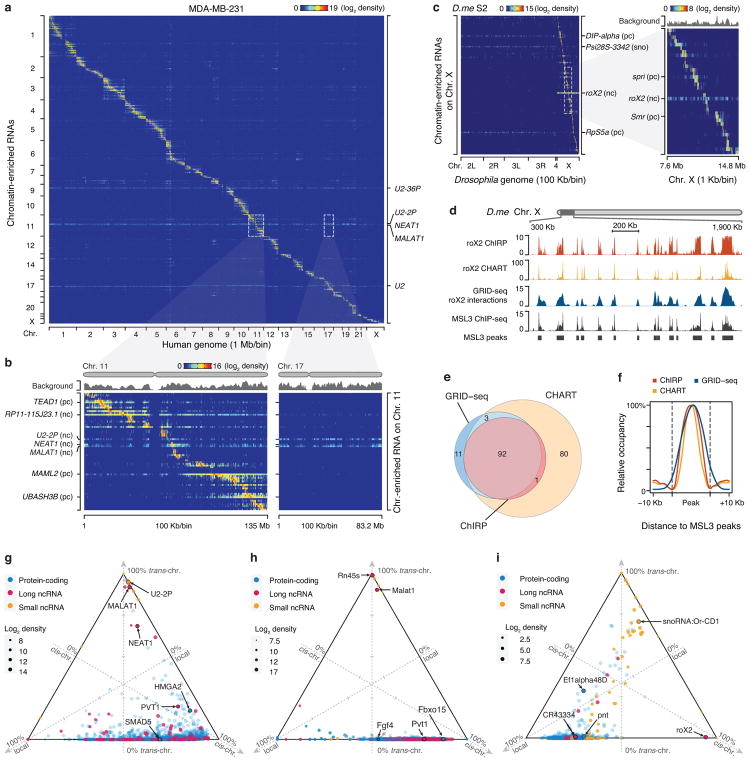
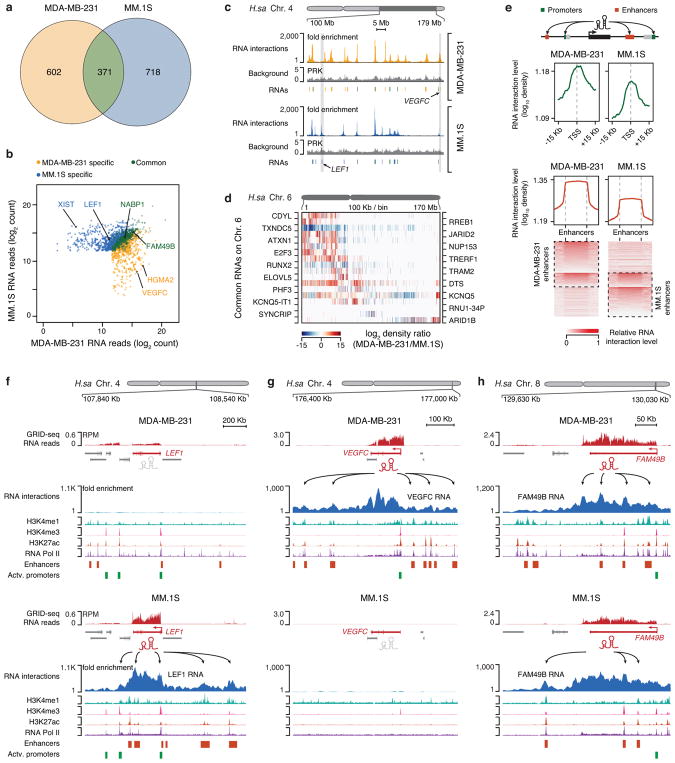
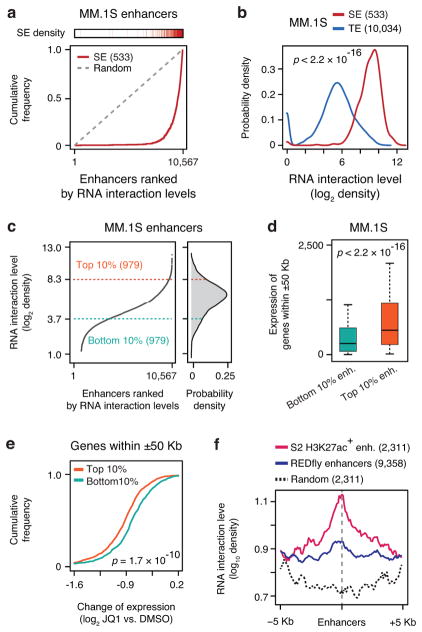
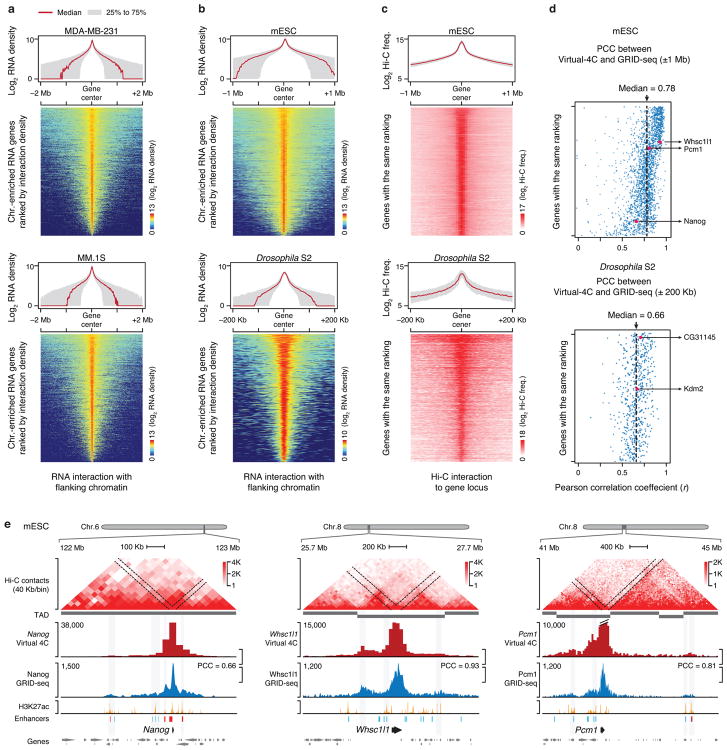
Higher eukaryotic genomes are bound by a large number of coding and non-coding RNAs, but approaches to comprehensively map the identity and binding sites of these RNAs are lacking. Here we report a method to capture in situ global RNA interactions with DNA by deep sequencing (GRID-seq), which enables the comprehensive identification of the entire repertoire of chromatin-interacting RNAs and their respective binding sites. In human, mouse, and Drosophila cells, we detected a large set of tissue-specific coding and non-coding RNAs that are bound to active promoters and enhancers, especially super-enhancers. Assuming that most mRNA-chromatin interactions indicate the physical proximity of a promoter and an enhancer, we constructed a three-dimensional global connectivity map of promoters and enhancers, revealing transcription-activity-linked genomic interactions in the nucleus.
高等真核生物基因组与大量编码和非编码RNA结合,但缺乏全面绘制这些RNA的身份和结合位点的方法。在此,我们报告了一种通过深度测序原位捕获RNA与DNA全局相互作用的方法(GRID-seq),该方法能够全面鉴定与染色质相互作用的RNA及其各自的结合位点。在人、小鼠和果蝇细胞中,我们检测到大量与活性启动子和增强子,尤其是超级增强子结合的组织特异性编码和非编码RNA。假设大多数mRNA-染色质相互作用表明启动子和增强子在物理上接近,我们构建了启动子和增强子的三维全局连接图谱,揭示了细胞核中与转录活性相关的基因组相互作用。