Institute of Technology, University of Washington, Tacoma Campus, Box 358426, 1900 Commerce Street, Tacoma, WA 98402-3100, U.S.A.
Department of Statistics, University of Washington, Box 354322, Seattle, WA 98195-4322, U.S.A.
Gigascience. 2017 Oct 1;6(10):1-10. doi: 10.1093/gigascience/gix078.
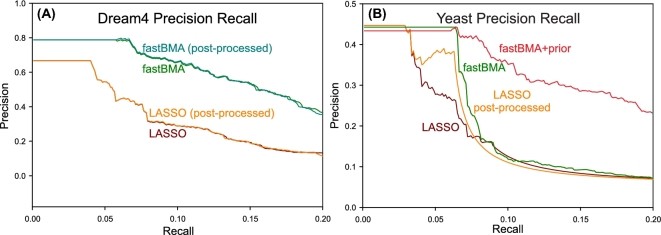
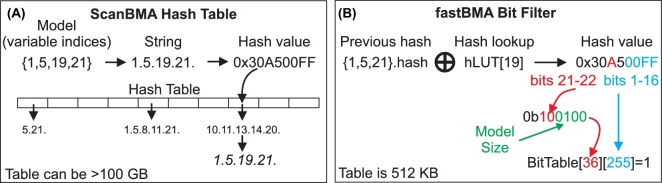
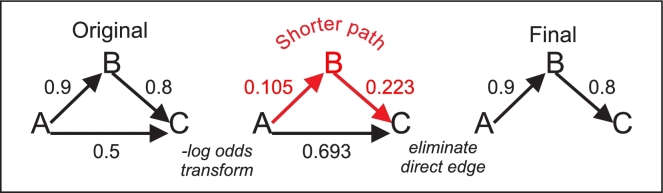
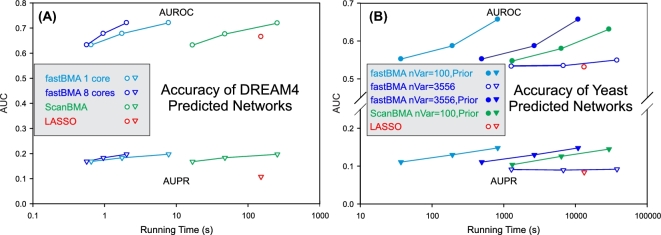
Inferring genetic networks from genome-wide expression data is extremely demanding computationally. We have developed fastBMA, a distributed, parallel, and scalable implementation of Bayesian model averaging (BMA) for this purpose. fastBMA also includes a computationally efficient module for eliminating redundant indirect edges in the network by mapping the transitive reduction to an easily solved shortest-path problem. We evaluated the performance of fastBMA on synthetic data and experimental genome-wide time series yeast and human datasets. When using a single CPU core, fastBMA is up to 100 times faster than the next fastest method, LASSO, with increased accuracy. It is a memory-efficient, parallel, and distributed application that scales to human genome-wide expression data. A 10 000-gene regulation network can be obtained in a matter of hours using a 32-core cloud cluster (2 nodes of 16 cores). fastBMA is a significant improvement over its predecessor ScanBMA. It is more accurate and orders of magnitude faster than other fast network inference methods such as the 1 based on LASSO. The improved scalability allows it to calculate networks from genome scale data in a reasonable time frame. The transitive reduction method can improve accuracy in denser networks. fastBMA is available as code (M.I.T. license) from GitHub (https://github.com/lhhunghimself/fastBMA), as part of the updated networkBMA Bioconductor package (https://www.bioconductor.org/packages/release/bioc/html/networkBMA.html) and as ready-to-deploy Docker images (https://hub.docker.com/r/biodepot/fastbma/).
从全基因组表达数据推断遗传网络在计算上要求极高。为此,我们开发了 fastBMA,这是一种用于贝叶斯模型平均(BMA)的分布式、并行和可扩展实现。fastBMA 还包括一个计算效率高的模块,通过将传递约简映射到易于解决的最短路径问题,来消除网络中的冗余间接边。我们在合成数据以及实验性全基因组时间序列酵母和人类数据集上评估了 fastBMA 的性能。使用单个 CPU 内核时,fastBMA 的速度比下一个最快的方法 LASSO 快 100 倍,准确性更高。它是一种内存高效、并行和分布式应用程序,可以扩展到人类全基因组表达数据。使用 32 核云集群(2 个 16 核节点),可以在几个小时内获得 10000 个基因调控网络。fastBMA 是对其前身 ScanBMA 的重大改进。它比其他快速网络推断方法(如基于 LASSO 的方法)更准确,速度快几个数量级。改进的可扩展性使其能够在合理的时间内计算来自基因组规模数据的网络。传递约简方法可以提高密集网络的准确性。fastBMA 可从 GitHub(https://github.com/lhhunghimself/fastBMA)获得代码(麻省理工学院许可证),作为更新的 networkBMA Bioconductor 包(https://www.bioconductor.org/packages/release/bioc/html/networkBMA.html)的一部分,并作为可立即部署的 Docker 映像(https://hub.docker.com/r/biodepot/fastbma/)。