Frolova Alina, Wilczyński Bartek
Institute of Molecular Biology and Genetics, Kyiv, Ukraine.
Institute of Informatics, University of Warsaw, Warsaw, Poland.
PeerJ. 2018 Oct 19;6:e5692. doi: 10.7717/peerj.5692. eCollection 2018.
Bayesian networks are directed acyclic graphical models widely used to represent the probabilistic relationships between random variables. They have been applied in various biological contexts, including gene regulatory networks and protein-protein interactions inference. Generally, learning Bayesian networks from experimental data is NP-hard, leading to widespread use of heuristic search methods giving suboptimal results. However, in cases when the acyclicity of the graph can be externally ensured, it is possible to find the optimal network in polynomial time. While our previously developed tool BNFinder implements polynomial time algorithm, reconstructing networks with the large amount of experimental data still leads to computations on single CPU growing exceedingly.
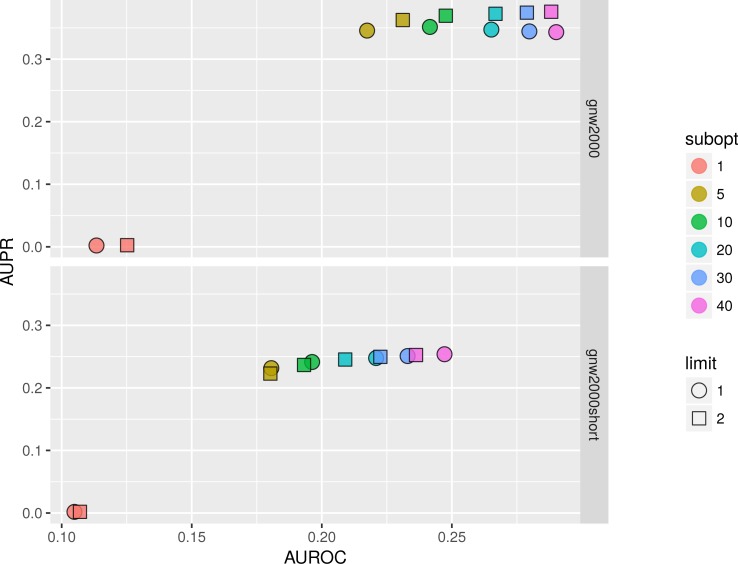
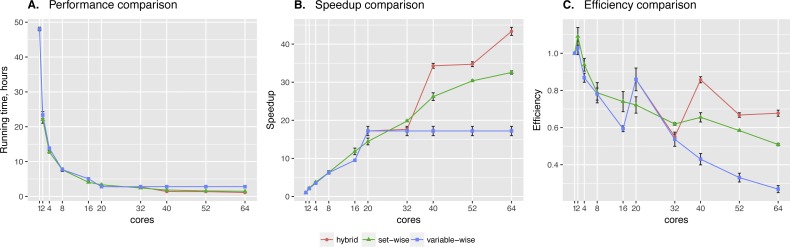
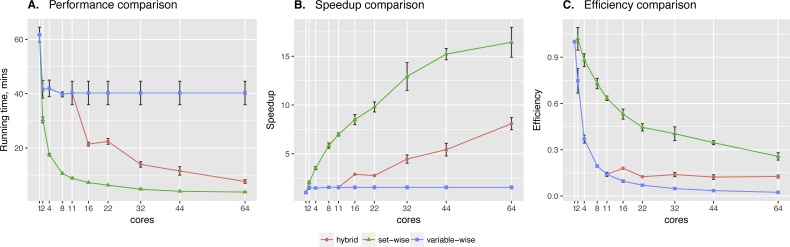
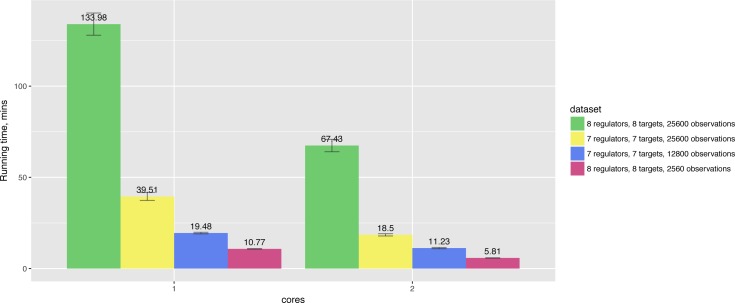
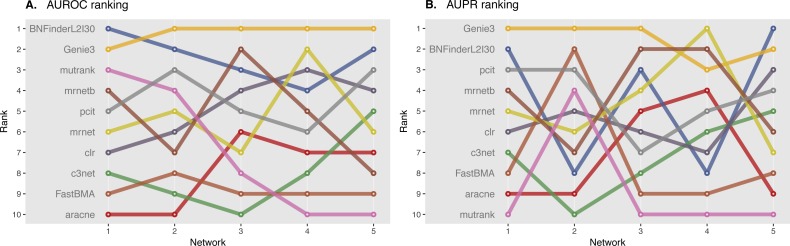
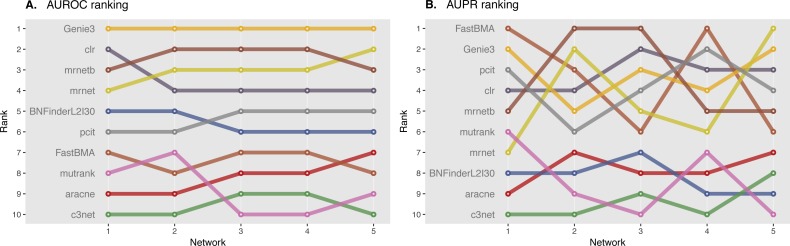
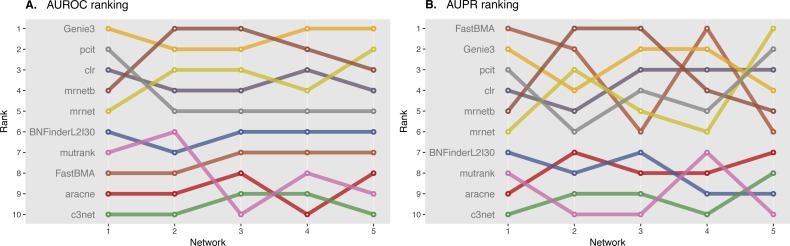
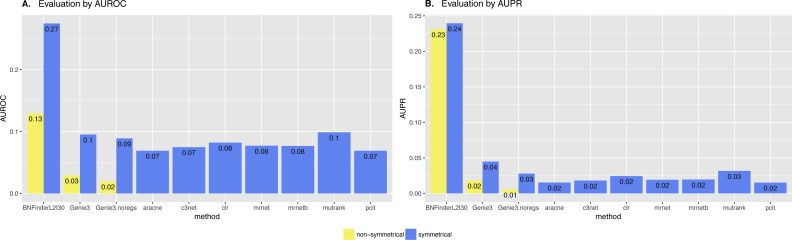
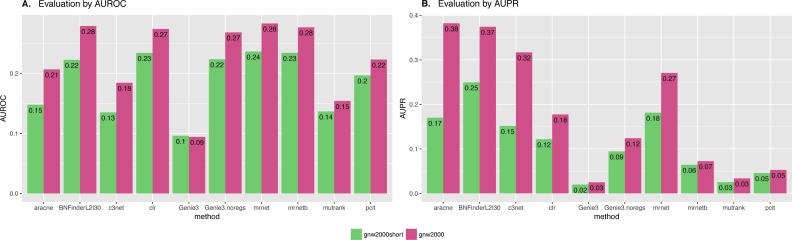
In the present paper we propose parallelized algorithm designed for multi-core and distributed systems and its implementation in the improved version of BNFinder-tool for learning optimal Bayesian networks. The new algorithm has been tested on different simulated and experimental datasets showing that it has much better efficiency of parallelization than the previous version. BNFinder gives comparable results in terms of accuracy with respect to current state-of-the-art inference methods, giving significant advantage in cases when external information such as regulators list or prior edge probability can be introduced, particularly for datasets with static gene expression observations.
We show that the new method can be used to reconstruct networks in the size range of thousands of genes making it practically applicable to whole genome datasets of prokaryotic systems and large components of eukaryotic genomes. Our benchmarking results on realistic datasets indicate that the tool should be useful to a wide audience of researchers interested in discovering dependencies in their large-scale transcriptomic datasets.
贝叶斯网络是一种有向无环图模型,广泛用于表示随机变量之间的概率关系。它们已被应用于各种生物学背景中,包括基因调控网络和蛋白质 - 蛋白质相互作用推断。一般来说,从实验数据中学习贝叶斯网络是NP难问题,这导致启发式搜索方法被广泛使用,但结果往往次优。然而,在图的无环性可以从外部确保的情况下,有可能在多项式时间内找到最优网络。虽然我们之前开发的工具BNFinder实现了多项式时间算法,但用大量实验数据重建网络仍然导致在单个CPU上的计算量急剧增加。
在本文中,我们提出了一种为多核和分布式系统设计的并行算法,并在改进版的BNFinder工具中实现,用于学习最优贝叶斯网络。新算法已在不同的模拟和实验数据集上进行了测试,结果表明它比以前的版本具有更好的并行效率。在准确性方面,BNFinder与当前最先进的推断方法给出了可比的结果,在可以引入诸如调节因子列表或先验边概率等外部信息的情况下具有显著优势,特别是对于具有静态基因表达观测值的数据集。
我们表明,新方法可用于重建数千个基因规模的网络,使其实际适用于原核系统的全基因组数据集以及真核基因组的大部分区域。我们在实际数据集上的基准测试结果表明,该工具对于广大有兴趣在其大规模转录组数据集中发现依赖性的研究人员应是有用的。