Sorbonne Universités, UPMC-Univ P6, CNRS, IBPS, Laboratoire de Biologie Computationnelle et Quantitative-UMR 7238, 75005 Paris, France.
Institut Universitaire de France, 75005 Paris, France.
Bioinformatics. 2018 Feb 1;34(3):459-468. doi: 10.1093/bioinformatics/btx584.
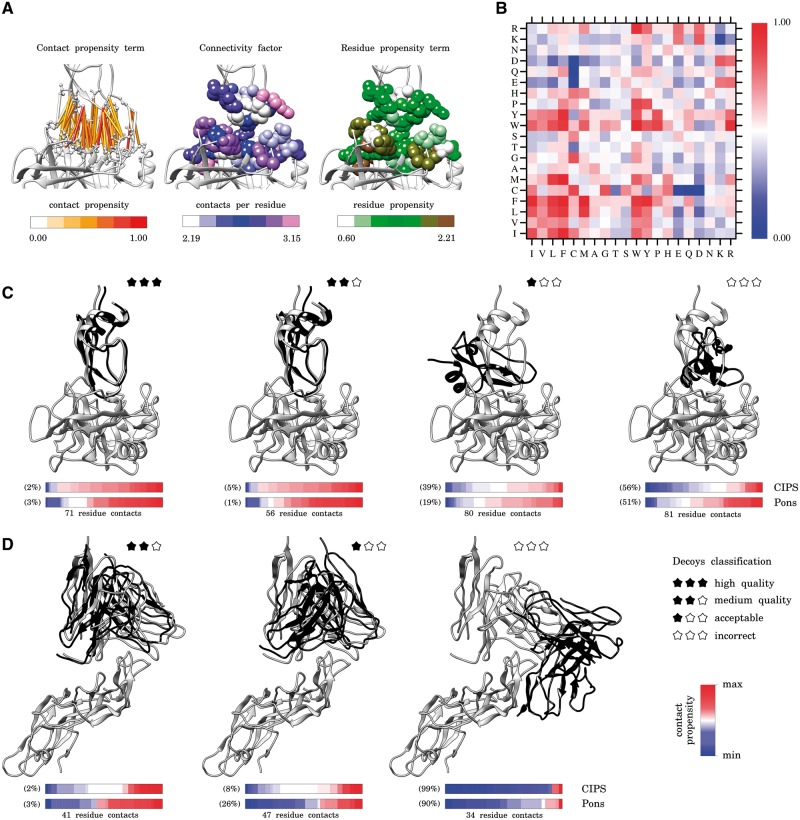
Large-scale computational docking will be increasingly used in future years to discriminate protein-protein interactions at the residue resolution. Complete cross-docking experiments make in silico reconstruction of protein-protein interaction networks a feasible goal. They ask for efficient and accurate screening of the millions structural conformations issued by the calculations.
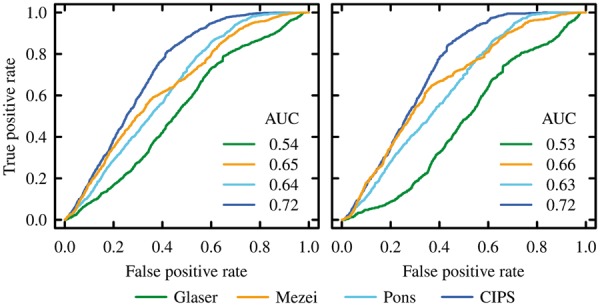
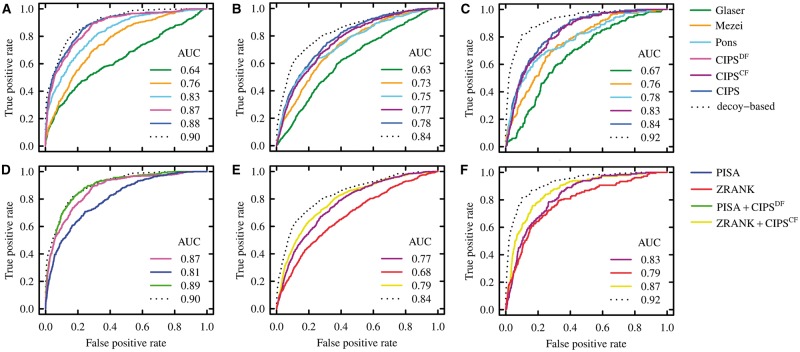
We propose CIPS (Combined Interface Propensity for decoy Scoring), a new pair potential combining interface composition with residue-residue contact preference. CIPS outperforms several other methods on screening docking solutions obtained either with all-atom or with coarse-grain rigid docking. Further testing on 28 CAPRI targets corroborates CIPS predictive power over existing methods. By combining CIPS with atomic potentials, discrimination of correct conformations in all-atom structures reaches optimal accuracy. The drastic reduction of candidate solutions produced by thousands of proteins docked against each other makes large-scale docking accessible to analysis.
CIPS source code is freely available at http://www.lcqb.upmc.fr/CIPS.
Supplementary data are available at Bioinformatics online.
未来几年,大规模的计算对接将越来越多地用于区分残基分辨率的蛋白质-蛋白质相互作用。完整的对接实验使蛋白质-蛋白质相互作用网络的计算重建成为一个可行的目标。它们需要有效地筛选出计算得出的数百万种结构构象。
我们提出了 CIPS(结合界面倾向用于诱饵评分),这是一种新的对势,将界面组成与残基-残基接触偏好结合在一起。CIPS 在筛选全原子或粗粒度刚性对接获得的对接解决方案方面优于其他几种方法。对 28 个 CAPRI 目标的进一步测试证实了 CIPS 相对于现有方法的预测能力。通过将 CIPS 与原子势相结合,可以达到全原子结构中正确构象的最佳区分精度。通过对数千种蛋白质相互对接产生的候选解决方案进行大量减少,使得大规模对接能够进行分析。
CIPS 的源代码可在 http://www.lcqb.upmc.fr/CIPS 上免费获得。
补充数据可在 Bioinformatics 在线获得。